
无菌粉末注射剂生产中可见异物检查的风险评估及消减措施
2014-10-10 06:13吴耀卫
机电信息 2014年8期
吴耀卫
(上海新亚药业有限公司,上海201203)
0 引言
2010版《中华人民共和国药典(二部)》附录IB注射剂中将注射剂定义为[1]:注射剂系指药物与适宜的溶剂或分散介质制成的供注入体内的溶液、乳状液或混悬液及供临用前配制或稀释成溶液或混悬液的粉末或浓溶液的无菌制剂。其分为注射液、注射用无菌粉末与注射用浓溶液。同时,药典明确规定:除另有规定外,注射剂应进行以下相应检查,即装量、装量差异、渗透压摩尔浓度、可见异物、无菌、细菌内毒素或热原。
随着对药品质量的不断提高,人们在注射剂中检出可见异物的几率越来越高,实际生产与质量要求之间的矛盾也相应凸现,这引起了人们的广泛关注。尤其在检测依据、控制判断标准、检测方式、生产过程中的防患措施等方面更是成为热门话题[2]。然而,无菌粉末注射剂生产中的可见异物在人、机、料、法、环各环节均存在,如何加强监控是保证制药质量的一大课题。本文将以抗生素类无菌粉末注射剂为例,探讨无菌粉末注射剂生产中的可见异物检查问题。
1 可见异物的定义与中国药典对其要求
1.1 可见异物的定义
在2010版中国药典二部附录IX H中将可见异物定义为:可见异物系指存在于注射剂、眼用液体制剂中,在规定条件下目视可以观测到的不溶性物质,其粒径或长度通常大于50μm。
1.2 中国药典对可见异物的要求
药典规定:不溶性物质粒径或长度通常≥50μm,即人工目视在规定的条件下或用自动灯检机检查时粒径或长度≥50μm的不溶性物才算可见异物。
1.3 可见异物的结果判定
药典规定:在静置一定时间后轻轻地旋转时均不得检出烟雾状微粒柱,且不得检出金属屑、玻璃屑、长度或最大粒径超过2 mm的纤维和块状物等明显可见异物。微细可见异物(如点状物、2 mm以下的短纤维和块状物等)如有检出,除另有规定外,应分别符合相应规定。
对注射用无菌粉末而言,被检查的5支(瓶)供试品中,均不得检出明显可见异物。若检出微细可见异物,每支(瓶)供试品中检出微细可见异物的数量应符合注射用无菌粉末微细可见异物限度(表1)的规定;若有1支(瓶)不符合规定,另取10支(瓶)同法复试,均应符合规定。
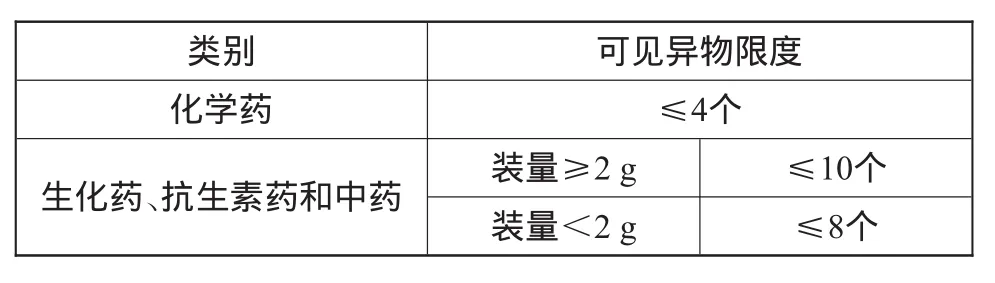
表1 注射用无菌粉末微细可见异物限度
然而,针对与粉针剂分装相关的无菌原料药,药典又规定:5份被检查的供试品中,均不得检出明显可见异物。若检出微细可见异物,每份供试品中检出微细可见异物的数量应符合无菌原料药微细可见异物限度(表2)的规定;若有1份不符合规定,另取10份同法复试,均应符合规定。
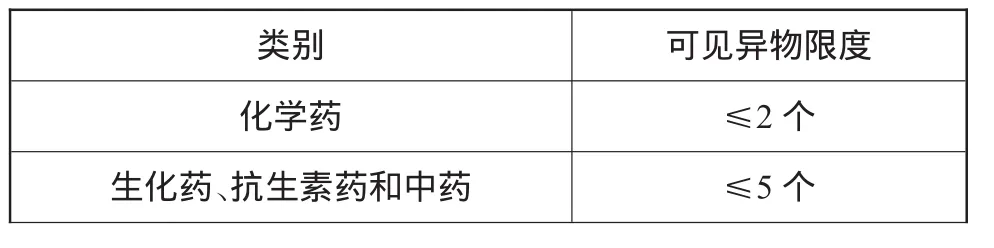
表2 无菌原料药微细可见异物限度
2 可见异物种类及产生原因
存在于注射剂中目视可常见的可见异物可以分为下列几种:金属类可见异物、玻璃类可见异物、纤维类可见异物和其他可见异物[2]。
以抗生素类无菌粉末注射剂为例,可见异物分类及产生原因如表3所示。
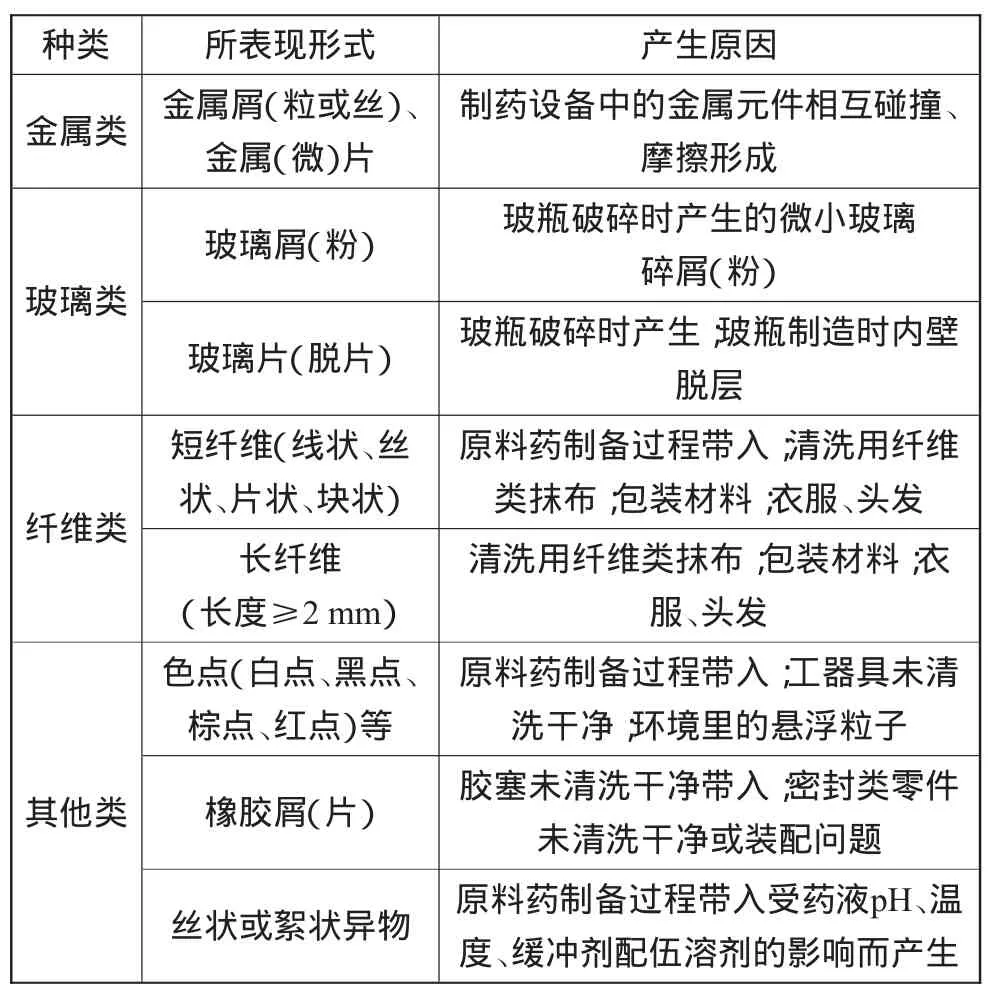
表3 可见异物分类及产生原因
3 可见异物的风险评估
3.1 从“人、机、料、法、环”分析可见异物
以抗生素类无菌粉末注射剂为例,药典要求检查其可见异物是以供试品为切入点,而抗生素类无菌粉末注射剂供试品一般为成品,即经分装、压塞与轧盖后,且含瓶子、胶塞、粉体与铝盖。因此,分析抗生素类无菌粉末注射剂可见异物的产生原因可结合使用鱼骨图分析法,综合归纳为“人、机、料、法、环”5个方面(表4)。
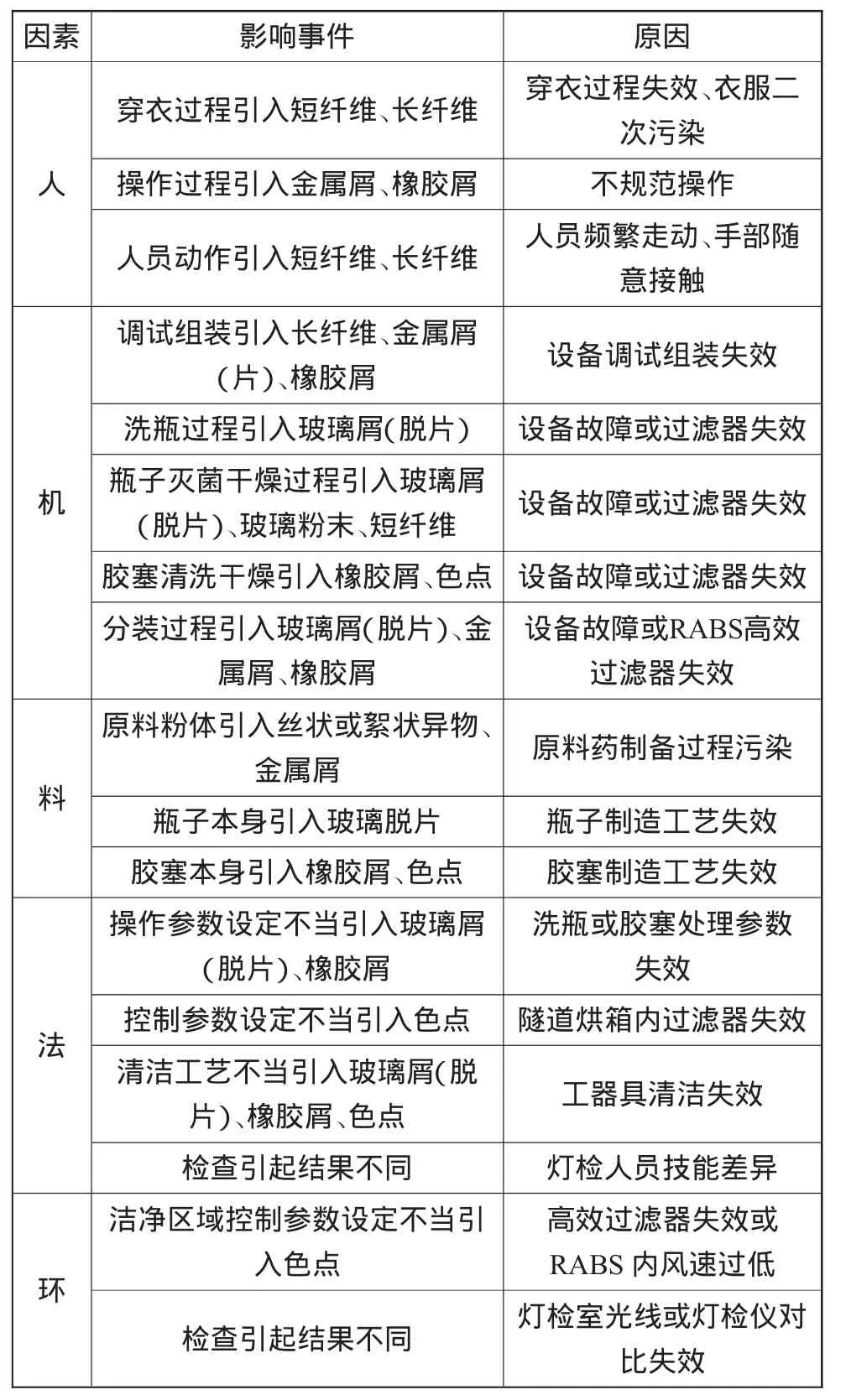
表4 抗生素类无菌粉末注射剂可见异物的发生原因
以上统计是造成抗生素类无菌粉末注射剂可见异物的发生原因汇总,现对其进行风险评估。
3.2 用FMEA法对可见异物进行风险评估
质量风险评估必须依靠相应的定性或定量的风险分析工具,常见工具包括失败模式与效应分析(FMEA)、故障树(FTA)、危害与可操作性分析(HAZOP)、危害分析和关键环节控制点(HACCP)等[3]。其中,FMEA较为常用,下述以FMEA法对可见异物进行风险评估。
3.2.1 风险评估方法简介
(1)风险事件识别。本步骤是对系统功能或子功能可能导致相关风险的风险事件进行识别,并确定事件所产生的影响。
(2)风险发生概率的评判。在确定所有可能发生的风险事件及其影响后,应对这些风险事件发生的概率进行判断。概率判断的标准并无统一规定,具体可根据事件的复杂程度来决定,现风险发生概率(L)的判断标准如表5所示。

表5 风险发生概率的判断标准
(3)风险事件严重性的评估。风险事件影响的评估应当全面,不仅需评估风险的直接影响,还应评估这些影响对于企业的长期和广泛影响。严重性判断的标准并无统一规定,现风险事件严重性(S)的判断标准如表6所示。
(4)风险等级的判定。风险等级的判定可以通过可见异物风险评估(表7)进行。在矩阵中,风险等级R=发生概率(L)×事件严重性(S)。
判定:风险等级R=20~25为巨大风险;R=12~16为重大风险;R=4~10为一般风险;R<4为轻微或可忽略的风险。
3.2.2 可见异物风险评估
可见异物风险评估如表7所示。
4 可见异物检查指标的建立与风险消减措施
从表7可以看出,子问题2.2、2.4为风险巨大,子问题3.3、4.1为风险重大,这几个风验是与可见异物相关且需重点考虑的问题。然而,这些风险巨大或重大问题归集起来便是洗瓶与胶塞清洗干燥工序问题,如何消减此类风险呢?对于抗生素类无菌粉末注射剂生产而言,关键是可见异物检查指标与风险消减措施的建立。
4.1 可见异物检查指标的建立
对于抗生素类无菌粉末注射剂的制剂生产而言,药典要求是检查供试品,供试品将由抗生素瓶、原料粉末与胶塞等组成,但此类总组成的微细可见异物限度为≤8个。然而,其中与粉针剂分装相关的无菌原料药,药典又规定微细可见异物限度为≤5个。因此,抗生素类无菌粉末注射剂生产环节的微细可见异物限度只能≤3个,这对于制剂生产过程来说无疑是一件困难的事。
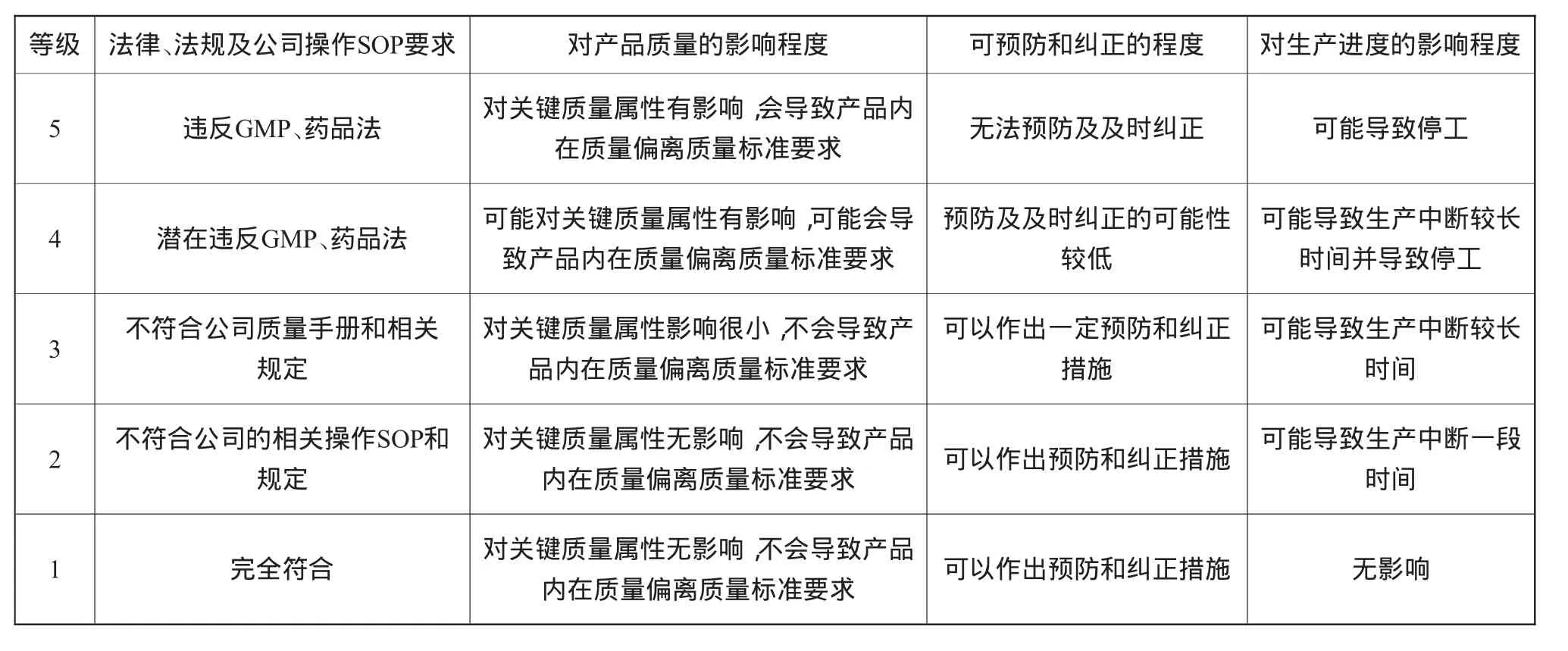
表6 风险事件严重性的判断标准
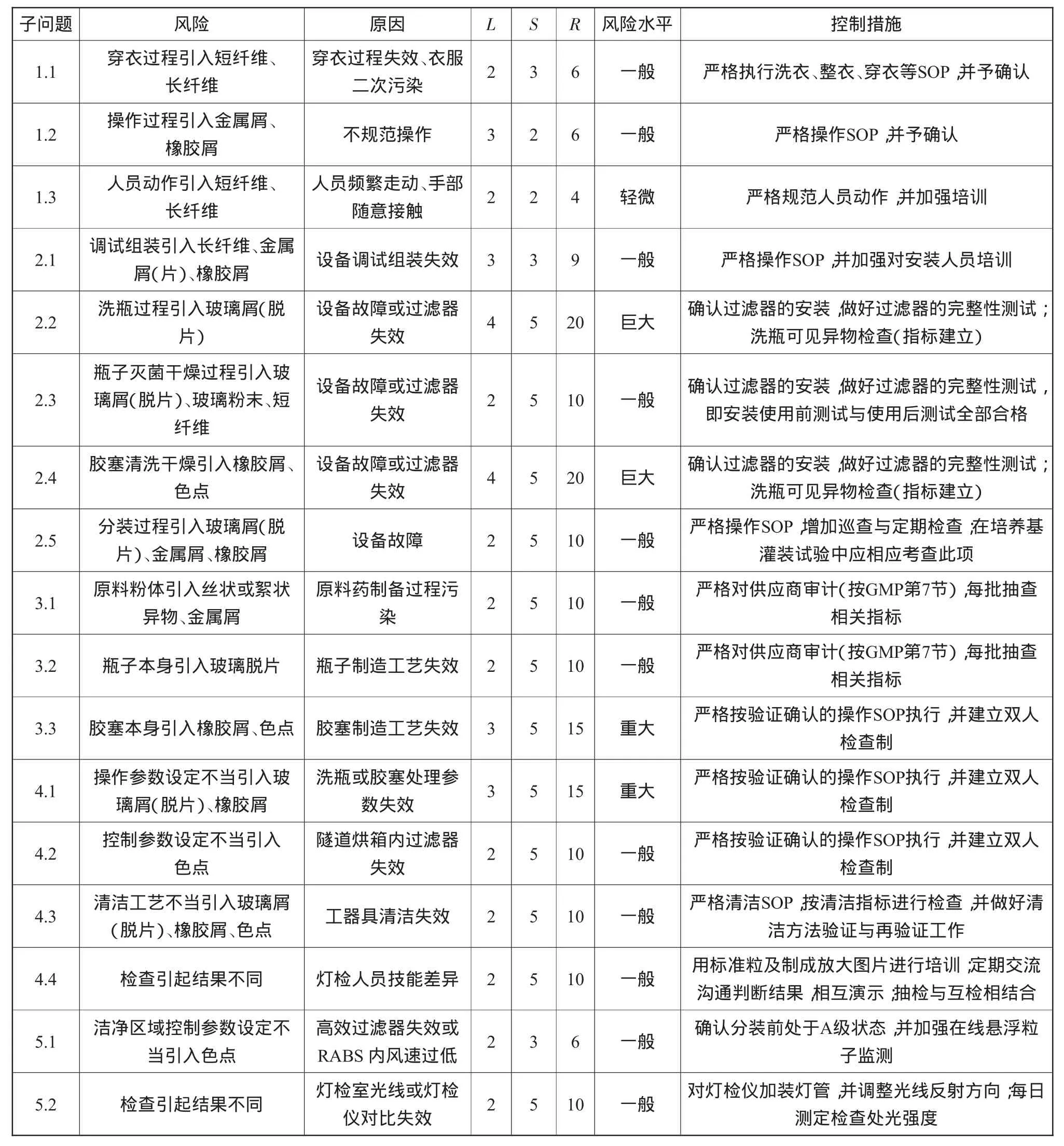
表7 可见异物风险评估
在寻找建立洗瓶与胶塞清洗干燥工序可见异物检查企标的理念依据时,我们在调研近3年多个无菌原料药供应商产品时发现,其抽检可见异物数量结果平均为2.33个,且最大可见异物数量为3个。为此,对无菌原料药粉末可见异物设置警戒限和行动限,警戒限为≤3个,行动限为≤4个。在此基础上,洗瓶与胶塞清洗干燥工序可见异物检查的企标就可方便建立,即抗生素瓶清洗后微细可见异物限度为≤2个,胶塞清洗干燥后微细可见异物限度为≤2个。同时,由于现生产均在RABS隔离下生产,只有严格控制其余环节的微细可见异物限度,才能使其消减数量在1个以下或无,这样才能使抗生素类无菌粉末注射剂生产的最终成品达到药典要求。
4.2 风险消减措施的建立
4.2.1 严格控制抗生素瓶与胶塞的审计与抽查
抗生素瓶审计与抽查要点:(1)在玻瓶制造过程中混入可见异物,如原辅材料、生产环境中的灰尘、清洁用抹布、破损的玻璃等;(2)在玻瓶质量控制过程中的易产生可见异物,如玻瓶壁上细微裂纹开裂形成的碎屑;(3)在玻瓶原料配方不当时易产生可见异物,如硼硅含量不当时会引起玻瓶内壁脱片。
胶塞审计与抽查要点:(1)选择与药物不相溶的橡胶制造胶塞,如卤化丁基胶、特种橡胶(共聚物);(2)胶塞生产厂房应符合GMP要求,在洁净区域生产并事先清洗干净,选用洁净的辅料与包装材料(密封包装),可有效减少橡胶屑的产生;(3)对于抗生素(头孢类)产品宜选用四氟乙烯覆膜丁基胶塞,可有效防止异物的产生。
4.2.2 加强检查人员的培训与考核
可见异物的检查受人为因素的影响较大,尤其是人工目视灯检法。其主要因素及消减措施如表8所示[2]。
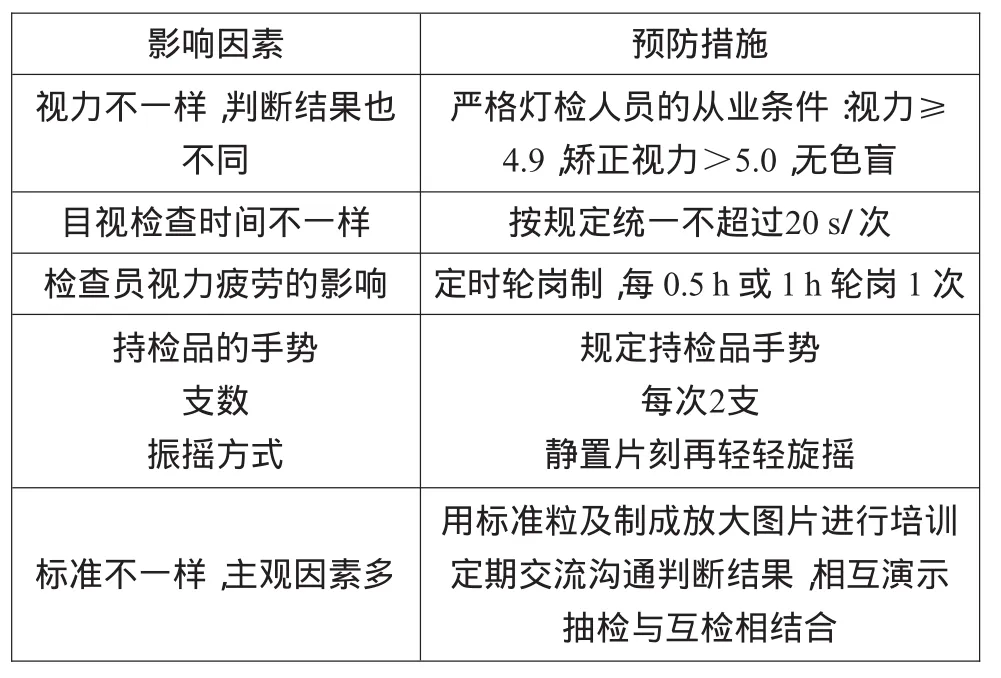
表8 检查人员对可见异物的影响因素及消减措施
4.2.3 与RABS技术结合可有效消减
现在设备均应用RABS技术,其能防止可见异物的混入,杜绝环境和人员对产品的污染,因为抗生素类无菌粉末注射剂生产时微生物的污染和可见异物的混入同时存在。同时,物料转移的隔离化与自净区域的设置也能有效地消减外界微生物和可见异物对药品的污染,确保药品质量的安全性。
4.2.4 文件制度的完善与严格执行
(1)建立设备与相关零部件清洗的SOP,建立容器具清洗的SOP,保证清洗人员规范操作。
(2)建立设备操作的SOP,特别是组装调试后的验收要求、过滤器性能监控。
4.2.5 其他消减措施
(1)人员培训。生产人员需经过严格的培训和考核,拿到合格证后方可上岗。
(2)生产安排的强度应适中,防止人员因过度疲劳导致操作不当等风险的发生。
(3)清洁验证及再验证。每个产品都要做清洁方法的适用性验证,即清洁验证,以所有产品的合格标准中的最低值为验证标准。
5 结语
本文从可见异物的定义与中国药典对其要求入手,以抗生素类无菌粉末注射剂为例,分析了可见异物种类及产生原因,继而对其进行风险评估,并探讨了可见异物检查指标的建立与风险消减的相关措施。由此可以得出结论:只有通过风险评估,强化有效消减风险措施,特别是相关工序检查企标的建立,才能使抗生素类无菌粉末注射剂的可见异物指标达到药典要求。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010
[2]周立法,赵小英.刍议注射剂“可见异物检查”[J].机电信息,2012(2)
[3]王燕,肖潇,梁毅.浅析质量风险管理在计算机化系统验证中的应用[J].机电信息,2011(4)
猜你喜欢
中老年保健(2022年4期)2022-08-22
天然气工业(2018年11期)2018-12-03
机电信息(2018年14期)2018-05-14
中成药(2018年2期)2018-05-09
中成药(2016年8期)2016-05-17
中国猪业(2016年1期)2016-01-28
中国中医药现代远程教育(2014年11期)2014-08-08
中国药业(2014年12期)2014-06-06
中国医药生物技术(2014年4期)2014-01-23
湖南畜牧兽医(2011年3期)2011-04-09
