
碱金属掺杂叔丁基杯[4]芳烃体系的结构及非线性光学性质
2014-09-21 08:59李志儒
物理化学学报 2014年7期
侯 娜 李 莹 吴 迪 李志儒
(吉林大学理论化学研究所,理论化学计算国家重点实验室,长春130023)
1 引言
在过去的30多年里,非线性光学(NLO)材料由于其在光通信、光信息存储、光计算及全光开关等高科技领域的潜在应用而引起科学工作者的重视.一阶超极化率作为非线性光学响应的一个重要标志受到了广泛关注.相关研究已报道了提高分子体系的一阶超极化率的多种策略,1-4碱金属掺杂是其中一个有效方法.5-10
碱金属化物(alkalide)是一种特殊的碱金属复合物晶体盐,其中碱金属阴离子占据了传统阴离子的位置.自从1974年Na+(cryptand[2.2.2])Na-首次被合成并表征,11,12具有独特电子结构和性质的碱金属化物就引起了人们的研究兴趣.近年,研究者将碱金属掺杂到各种配体中,理论设计或实验合成了很多种类的碱金属化物体系.此外,对一些碱金属化物分子的理论研究表明,它们具有非常大的非线性光学响应,是有潜力的非线性光学分子,如Li+(calix[4]pyrrole)M-(M=Li,Na,K)和 Li+(NH3)nNa-等.13,14
在上世纪四、五十年代,化学家Zinke等15-17发现了一种高熔点的环状四聚体.直到70年代末期,随着对冠醚、环糊精等大环化合物的研究工作的发展,这类环状四聚体引起了美国化学家Gutsche的兴趣和深入研究,并将其命名为杯芳烃.18其中,由对叔丁基苯酚和亚甲基单元聚合得到的环状四聚体称为叔丁基杯[4]芳烃(t-Bu-calix[4]arene),对这类体系的实验合成及其光谱性质等方面已有很多报道.19-22而对t-Bu-calix[4]arene的几何结构、振动性质等方面的理论研究也取得了一系列进展.23,24但迄今为止还没有对金属掺杂的t-Bu-calix[4]arene体系的理论或实验研究工作出现.之前的研究结果表明t-Bu-calix[4]arene具有四种构象,其中最稳定构型为锥型.在本课题组的工作中发现,具有类似锥型结构的calix[4]pyrrole分子,在掺杂碱金属原子后表现出大的非线性光学响应.13,25因此,我们以理论方法研究了t-Bu-calix[4]arene的碱金属掺杂效应.用密度泛函方法计算和对比讨论了t-Bu-calix[4]arene、单碱金属掺杂的M@t-Bu-calix[4]arene及双碱金属掺杂的(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的几何结构和非线性光学性质.希望我们的工作能为优良NLO材料的设计及合成提供理论依据.
2 计算方法
本文采用B3LYP方法,选取6-31G、6-31+G、6-31G(d)及6-31+G(d)基组对Li@t-Bu-calix[4]arene体系的最稳定结构进行优化测试,结果表明使用6-31G(d)和6-31+G(d)基组得到的优化结构参数几乎一致.因此,在B3LYP/6-31G(d)水平优化了M@t-Bu-calix[4]arene及(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的结构,并进行了频率计算,得到了全实频的几何构型.其中(M@t-Bu-calix[4]arene)Li'的基态结构是自旋单重态.使用同样的方法计算了体系的垂直电离能(VIE).使用B3LYP/6-31+G(d)方法计算了体系的自然键轨道(NBO)电荷.之前的研究表明CAM-B3LYP是一种适合用来计算碱金属掺杂体系的超极化率的密度泛函方法.26,27因此本文采用CAM-B3LYP方法计算了分子的平均偶极矩(μ0)、平均极化率(α0)和平均一阶超极化率(β0).在计算中对配体t-Bu-calix[4]arene的C和H原子使用6-31G基组,对氧原子及碱金属原子使用6-31+G(d)基组.对μ0,α0和β0的定义如下:
3 结果与讨论
3.1 几何结构及相对稳定性
图1中列出了单碱金属掺杂的M@t-Bu-calix[4]arene(M=Li,K)体系的全实频几何结构,以及最高占据分子轨道.如图所示,Li@t-Bu-calix[4]arene与K@t-Bu-calix[4]arene体系各有两个异构体,在Li@t-Bu-calix[4]arene的最低能量结构A和K@t-Bu-calix[4]arene的次稳定异构体B中,碱金属原子处于t-Bu-calix[4]arene配体内部接近一个亚甲基的位置.而Li@t-Bu-calix[4]arene的异构体B和K@t-Bu-calix[4]arene的最稳定异构体A中碱金属原子处于配体的C4轴上.Na@t-Bu-calix[4]arene体系只有一个稳定结构,与K@t-Bu-calix[4]arene的异构体A结构相似.这些异构体的相对稳定性与掺杂原子的尺寸有关,当掺杂的碱金属原子(Li)较小时,为了和配体更好地结合,它倾向于偏向配体的一边;而掺杂的碱金属为较大的Na和K时,它们则倾向于配体的中心位置.而对于杯[4]吡咯分子而言,掺杂一个锂原子后的Li@calix[4]pyrrole体系只有一个类似于Li@t-Bu-calix[4]arene的异构体B的稳定结构,25这是由于杯[4]吡咯的尺寸较小,Li原子位于配体中心就可以达到和它的最优结合.此外,通过对比t-Bu-calix[4]arene和M@t-Bu-calix[4]arene体系中对角亚甲基之间的距离,发现单碱金属掺杂对于t-Bu-calix[4]arene分子的结构几乎没有影响.
在M@t-Bu-calix[4]arene的稳定结构的基础上,我们在配体外部又掺杂了一个Li原子,并考虑了各种可能的初始结构,经过几何优化和频率计算得到了稳定的异构体.图2中列出了(M@t-Bu-calix[4]arene)Li'体系最低能量结构MLi'-1(M=Li,Na,K)及其它低能级异构体,表1列出了主要的结构参数,其中d(M-Li')代表配体内外碱金属之间的距离,d(MO)和d(Li'-O)值代表掺杂的碱金属原子与图2中所标示出的配体底部氧原子之间的距离.如图2所示,每个双碱金属掺杂的(M@t-Bu-calix[4]arene)Li'体系有5个稳定异构体.在(M@t-Bu-calix[4]arene)Li'的最稳定结构LiLi'-1、NaLi'-1和KLi'-1中,Li'原子都偏向t-Bu-calix[4]arene配体的一侧.在LiLi'-1结构中,Li原子几乎处于四个O原子的中心位置,而在NaLi'-1和KLi'-1结构中,Na和K原子位于t-Bucalix[4]arene配体的内部中心位置附近.在MLi'-2、MLi'-3、MLi'-4结构中,配体内部的M原子偏离笼中心位置.异构体MLi'-2和MLi'-3的结构相似,M和Li'原子分别位于一个芳香环内外两侧,前者的Li'原子靠近配体底部的羟基,后者的Li'原子靠近芳香环的中心位置.在MLi'-4结构中M和Li'原子分别位于一个亚甲基内外两侧,Li'原子距离亚甲基较远.在异构体MLi'-5中M原子位于t-Bu-calix[4]arene配体的内部中心位置,而Li'原子与t-Bu-calix[4]arene配体底部的一个O原子非常接近.对比而言,(Li@calix[4]pyrrole)M(M=Li,Na,K)13体系只有一个稳定结构,说明对于尺寸较大的t-Bu-calix[4]arene配体,碱金属掺杂的位置更多样.
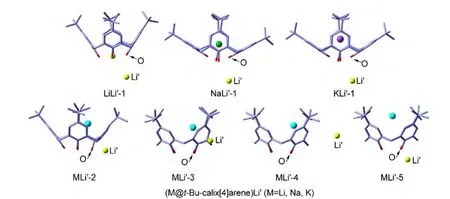
图2 (M@t-Bu-calix[4]arene)Li′(M=Li,Na,K)体系的优化几何结构Fig.2 Optimized structures of the(M@t-Bu-calix[4]arene)Li′(M=Li,Na,K)species

表1 (M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系各异构体的相对能量(Erel)、重要的原子间距离(d)及碱金属与配体之间的相互作用能(Eint)Table 1 Relative energies(Erel),important interatomic distances(d),and interaction energies(Eint)of the isomers of the(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)species
如图2所示,在(M@t-Bu-calix[4]arene)Li'体系的最低能量结构中,配体内碱金属M随原子序数的增大而逐渐远离t-Bu-calix[4]arene配体的底端,这一点可由表1中d(M-O)的变化趋势反映出来,即0.199 nm(LiLi'-1)<0.230 nm(NaLi'-1)<0.285 nm(KLi'-1).这一变化趋势导致体系中两个掺杂碱金属之间的距离d(M-Li')对M的原子序数的明显依赖性.由表1可见,d(M-Li')的变化顺序为0.387 nm(LiLi'-1)<0.538 nm(NaLi'-1)<0.604 nm(KLi'-1).
为了考查掺杂的碱金属与杯芳烃之间的结合力,我们使用Counterpoise方法计算了碱金属与t-Bu-calix[4]arene配体间的相互作用能.单掺杂的M@t-Bu-calix[4]arene(M=Li,Na,K)体系的相互作用能在0.67--30.96 kJ·mol-1之间,说明金属M与配体之间的相互作用较弱.对比来看,双碱金属掺杂的体系的前三个异构体的相互作用能很高,在-142.26--316.98 kJ·mol-1范围内,说明双金属掺杂更有利于体系的稳定.由表1可知,(M@t-Bucalix[4]arene)Li'的构型MLi'-4和MLi'-5的相互作用能基本都是正值,说明这两个异构体有分解的趋势,稳定性不及前三种更低能级的结构.
3.2 自然键轨道(NBO)电荷与垂直电离能(VIE)分析
单掺杂的M@t-Bu-calix[4]arene和双掺杂的(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系中碱金属上的NBO电荷列于表2中.图3给出了(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的HOMO图.如表中所示,Li@t-Bu-calix[4]arene的异构体A及K@t-Bu-calix[4]arene的异构体B中Li和K原子的电荷分别为0.757e和0.882e,说明Li和K原子掺杂到t-Bu-calix[4]arene分子中后失去了最外层价电子,以Li+和K+阳离子的形式存在.从其HOMO轨道图(图1)也可以看出碱金属的价电子转移到配体上.在M@t-Bu-calix[4]arene(M=Na,K)体系的最稳定结构及Li@t-Bu-calix[4]arene的次稳定结构内碱金属原子的NBO电荷在-0.067e-0.041e之间,接近于零,说明M和有机配体间几乎没有电荷转移,这和图1中这几个结构相对应的HOMO轨道图是一致的.
在(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的最低能量异构体中,内嵌金属M的NBO电荷在0.430e-0.681e之间,说明M失去一个电子成为M+阳离子.同时,外掺杂的Li'原子的NBO电荷均为负值,在-0.278e--0.444e范围内,表明Li'以阴离子的形式存在,因此整个体系显示出碱金属化物的电子特征.这一点在异构体MLi'-1的HOMO轨道中表现得尤为明显.由图3还可以看出,MLi'-4异构体内部掺杂的碱金属M的电子一部分转移给了相邻的芳香环,一部分转移到了外掺杂的Li'原子上.因此,异构体MLi'-4也包含着Li'-阴离子,是碱金属化物分子.异构体MLi'-2和MLi'-3中内嵌碱金属M和外掺Li'原子的电荷分别在0.545e-0.714e和0.562e-0.699e范围内变化,说明两个掺杂的碱金属都失去了最外层价电子,以阳离子形式存在.如图3所示,异构体MLi'-2和MLi'-3的HOMO轨道与文献30中的两个锂原子掺杂的bis(3,4-difluorophenyl)methane分子的LL结构的HOMO轨道相似,配体t-Bu-calix[4]arene上的芳香环对内部的碱金属M和外部的Li'都有显著的拉电子效应,说明M和Li'失去的电子转
移给了t-Bu-calix[4]arene分子.在LiLi'-5结构中,Li和Li'原子的NBO电荷分别为-0.189e和0.156e,可见通过配体t-Bu-calix[4]arene与碱金属的作用,内掺杂Li原子得到电子而外掺杂的Li'原子失去电子.类似地,在异构体NaLi'-5中,内掺杂的Na原子得到电子以阴离子形式存在.这两个碱金属化物分子的电子结构特点非常特殊,即碱金属阴离子存在于配体的内部位置.这一点和之前报道过的碱金属化物25,31都是不同的.

表2 (M@t-Bu-calix[4]arene)Li'、M@t-Bu-calix[4]arene(M=Li,Na,K)和t-Bu-calix[4]arene体系掺杂碱金属原子上的NBO电荷(q)、垂直电离能(VIE)、平均偶极矩(µ0)、极化率(α0)、一阶超极化率(β0)、最主要激发态的振子强度(f0)以及跃迁能(ΔE)Table 2 NBO charge of doping alkali atoms(q),vertical ionization energies(VIE),the mean dipole moments(µ0),polarizabilities(α0),first hyperpolarizabilities(β0),oscillator strength(f0)and transition energy(ΔE)of the crucial excited state of the(M@t-Bu-calix[4]arene)Li',M@t-Bu-calix[4]arene(M=Li,Na,K),and t-Bu-calix[4]arene species
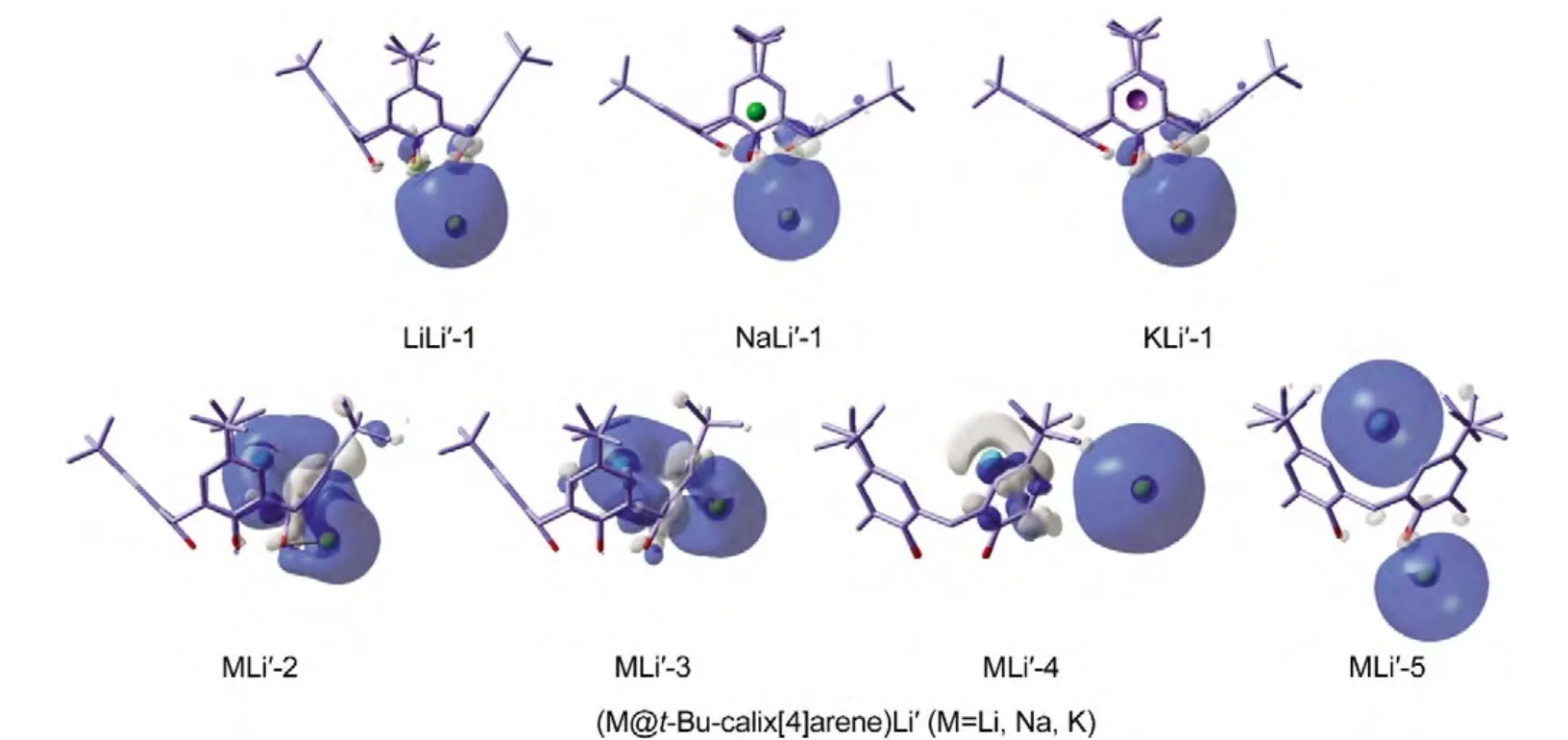
图3 (M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的HOMO图Fig.3 HOMO images of the(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)species
表2中列出了t-Bu-calix[4]arene、M@t-Bu-calix[4]arene和(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的垂直电离能(VIE)值.显然,碱金属掺杂使t-Bu-calix[4]arene体系的垂直电离能降低了50%左右.由表可见,在每个(M@t-Bu-calix[4]arene)Li'体系中,最稳定异构体的垂直电离能相对最高.对每个构型来说,由于外掺杂的碱金属原子都是Li,因此其VIE值主要由内掺杂的碱金属原子对电子的束缚能力决定.以构型MLi'-1为例,其垂直电离能随着内掺碱金属(M)的原子序数的递增而单调递减,即4.08 eV(M=Li)>3.89 eV(M=Na)>3.76 eV(M=K).这与M原子自身的电离能变化趋势是一致的.
3.3 非线性光学(NLO)性质
使用CAM-B3LYP方法计算了t-Bu-calix[4]arene、M@t-Bu-calix[4]arene和(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)体系的非线性光学性质,如表2所示.孤立的t-Bu-calix[4]arene分子的平均极化率为454 a.u.,通过对比可以发现碱金属掺杂提高了体系的平均极化率,其中双碱金属掺杂的MLi'-4和MLi'-5体系具有更大的平均极化率(890-1392 a.u.).同时计算结果表明在t-Bu-calix[4]arene分子中掺杂碱金属原子显著提高了体系的一阶超极化率.t-Bu-calix[4]arene的β0值仅为163 a.u.,掺杂一个碱金属原子后,β0为1105-29857 a.u.,再掺杂一个Li'原子后体系的β0达到了12719-114354 a.u..对于单掺杂的M@t-Bu-calix[4]arene体系而言,碱金属与t-Bu-calix[4]arene分子间发生电荷转移的异构体,即Li@t-Bu-calix[4]arene的结构A和K@t-Bu-calix[4]arene的结构B表现出更大的β0,分别为12302和29857 a.u..而其它异构体的β0相对较小,在1105-7087 a.u.之间.对于相同构型的M@t-Bu-calix[4]arene分子,其β0值对所掺杂的M原子存在着显著的依赖性,即β0随M的原子序数升高而增大.双碱金属掺杂的(M@t-Bu-calix[4]arene)Li'体系的异构体MLi'-4具有最高的β0,并且随着配体内部碱金属原子序数的增加而逐渐增大,即41827 a.u.(M=Li)<59777 a.u.(M=Na)<114354 a.u.(M=K).其中异构体KLi'-4的一阶超极化率是t-Bu-calix[4]arene分子的701倍,是单碱金属掺杂的K@t-Bu-calix[4]arene的异构体B的3倍.碱金属化物LiLi'-4和LiLi'-5的一阶超极化率β0分别为41827和33724 a.u.,比不同配体的碱金属化物(Li@calix[4]pyrrole)Li'体系25的β0大得多.从以上分析可以看出,在叔丁基杯[4]芳烃中引入碱金属原子可以极大地增强化合物的非线性光学响应,而且体系的一阶超极化率值受掺入的碱金属原子的个数及原子序数的影响.
为了分析碱金属掺杂后体系具有较大一阶超极化率的原因,我们可以参考静态条件下简化的双能级公式,32

上式中,f0、Δμ和ΔE分别代表体系最主要激发态的振子强度、基态与激发态之间的偶极矩差和跃迁能.式中一阶超极化率与ΔE的3次方成反比,可见ΔE是影响体系β0值的最主要因素.本文采用含时密度泛函理论(TD-DFT)的B3LYP方法计算得到了t-Bu-calix[4]arene、M@t-Bu-calix[4]arene和(M@t-Bucalix[4]arene)Li'(M=Li,Na,K)体系的最主要激发态的跃迁能和振子强度,相应的数据列于表2.对于孤立的t-Bu-calix[4]arene分子,其最主要激发态的跃迁能相对较高,为4.777 eV.而碱金属的掺杂带给了体系弥散的电子云(见图1和图3),这一特性使体系的基态电子很容易被激发,因此M@t-Bu-calix[4]arene和(M@t-Bu-calix[4]arene)Li'体系的最主要激发能很小,仅为0.941-2.789 eV,这是掺杂碱金属后体系具有很大的一阶超极化率的本质原因.
4 结论
报道了碱金属掺杂叔丁基杯[4]芳烃体系M@t-Bu-calix[4]arene和(M@t-Bu-calix[4]arene)Li'(M=Li,Na,K)的结构和性质.和其它不同配体的碱金属掺杂体系相比,这两种体系在结构方面表现出多样性的特点.NBO结果表明在部分(M@t-Bu-calix[4]arene)Li'复合物的异构体中,Li'原子以阴离子形式存在,整个体系表现出碱金属化物的特点.当t-Bucalix[4]arene内部掺杂一个碱金属原子(Li,Na,K)后,体系的一阶超极化率有较大提高,而在配体外部又掺杂一个锂原子后,体系表现出更大的β0值.可见,碱金属掺杂是提高t-Bu-calix[4]arene体系非线性光学响应的有效方法,而且体系的β0值对掺杂的碱金属数目及其原子序数均有一定依赖性.此工作通过对碱金属掺杂有机配体体系的系统研究,旨在为优良NLO材料的设计提供理论支持.
(1) Albert,I.D.L.;Marks,T.J.;Ratner,M.A.J.Am.Chem.Soc.1997,119,6575.doi:10.1021/ja962968u
(2) Coe,B.J.;Foxon,S.P.;Harper,E.C.;Raftery,J.;Shaw,R.;Swanson,C.A.;Asselberghs,I.;Clays,K.;Brunschwig,B.S.;Fitch,A.G.Inorg.Chem.2009,48,1370.doi:10.1021/ic801224u
(3) Karamanis,P.;Pouchan,C.J.Phys.Chem.C 2012,116,11808.doi:10.1021/jp3026573
(4) Janjua,M.R.S.A.;Liu,C.G.;Guan,W.;Zhuang,J.;Muhammad,S.;Yan,L.K.;Su,Z.M.J.Phys.Chem.A 2009,113,3576.doi:10.1021/jp808707q
(5) Xu,H.L.;Li,Z.R.;Wu,D.;Ma,F.;Li,Z.J.;Gu,F.L.J.Phys.Chem.C 2009,113,4984.doi:10.1021/jp806864w
(6)Muhammad,S.;Xu,H.L.;Liao,Y.;Kan,Y.H.;Su,Z.M.J.Am.Chem.Soc.2009,131,11833.doi:10.1021/ja9032023
(7) Jiang,Y.;Liu,Z.;Liu,H.;Cui,W.;Wang,N.;Liu,D.;Ge,X.Chin.Sci.Bull.2012,57,4448.
(8) Wang,L.J.;Sun,S.L.;Zhong,R.L.;Liu,Y.;Wang,D.L.;Wu,H.Q.;Xu,H.L.;Pan,X.M.;Su,Z.M.RSC Adv.2013,3,13348.doi:10.1039/c3ra40909k
(9) Wang,Y.F.;Huang,J.G.;Zhou,G.P.Struct.Chem.2013,24,1545.doi:10.1007/s11224-012-0188-7
(10) Fan,L.T.;Li,Y.;Wu,D.;Li,Z.R.;Sun,C.C.Acta Phys.-Chim.Sin.2012,28,555.[樊丽涛,李 莹,吴 迪,李志儒,孙家钟.物理化学学报,2012,28,555.]doi:10.3866/PKU.WHXB201112212
(11) Dye,J.L.;Ceraso,J.M.;Lok,M.;Barnett,B.;Tehan,F.J.J.Am.Chem.Soc.1974,96,608.doi:10.1021/ja00809a060
(12) Tehan,F.J.;Barnett,B.;Dye,J.L.J.Am.Chem.Soc.1974,96,7203.doi:10.1021/ja00830a005
(13) Chen,W.;Li,Z.R.;Wu,D.;Li,Y.;Sun,C.C.;Gu,F.L.;Aoki,Y.J.Am.Chem.Soc.2006,128,1072.doi:10.1021/ja056314+
(14) Jing,Y.Q.;Li,Z.R.;Wu,D.;Li,Y.;Wang,B.Q.;Gu,F.L.;Aoki,Y.ChemPhysChem 2006,7,1759.
(15) Zinke,A.;Ziegler,E.Ber.Dtsch.Chem.Ges.1944,77B,264.
(16) Zinke,A.;Kretz,R.;Leggewie,E.;Hössinger,K.Monatsh.Chem.1952,83,1213.doi:10.1007/BF00899467
(17) Ott,R.;Zinke,A.Oesterr.Chem.Ztg.1954,55,156
(18) Gutsche,C.D.;Muthukrishnan,R.J.Org.Chem.1978,43,4905.doi:10.1021/jo00419a052
(19) Gutsche,C.D.Accounts Chem.Res.1983,16,161.doi:10.1021/ar00089a003
(20) Gutsche,C.D.;Bauer,L.J.J.Am.Chem.Soc.1985,107,6052.doi:10.1021/ja00307a038
(21)Andreetti,G.D.;Pochini,A.;Ungaro,R.J.Chem.Soc.,Perkin Trans.2 1983,1773.
(22) Grootenhuis,P.D.J.;Kollman,P.A.;Groenen,L.C.;Reinhoudt,D.N.;Van Hummel,G.J.;Ugozzoli,F.;Andreetti,G.D.J.Am.Chem.Soc.1990,112,4165.doi:10.1021/ja00167a010
(23) Khedkar,J.K.;Pinjari,R.V.;Gejji,S.P.J.Phys.Chem.A 2011,115,10624.doi:10.1021/jp205441s
(24) Furer,V.;Borisoglebskaya,E.;Zverev,V.;Kovalenko,V.Spectrochim.Acta A 2006,63,207.doi:10.1016/j.saa.2005.05.006
(25) Chen,W.;Li,Z.R.;Wu,D.;Li,Y.;Sun,C.C.;Gu,F.L.J.Am.Chem.Soc.2005,127,10977.doi:10.1021/ja050601w
(26) Wang,J.J.;Zhou,Z.J.;Bai,Y.;Liu,Z.B.;Li,Y.;Wu,D.;Chen,W.;Li,Z.R.;Sun,C.C.J.Mater.Chem.2012,22,9652.doi:10.1039/c2jm15405f
(27) Karamanis,P.;Marchal,R.;Carbonniére,P.;Pouchan,C.J.Chem.Phys.2011,135,044511.doi:10.1063/1.3615499
(28) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,RevisionA02;Gaussian Inc.:Wallingford,CT,2009.
(29) Dennington,R.;Keith,T.;Millam,J.;et al.GaussView,version 5;Semichem Inc.:Shawnee Mission,KS,2009.
(30) Liu,Z.B.;Zhou,Z.J.;Li,Y.;Li,Z.R.;Wang,R.;Li,Q.Z.;Li,Y.;Jia,F.Y.;Wang,Y.F.;Li,Z.J.Phys.Chem.Chem.Phys.2010,12,10562.doi:10.1039/c004262e
(31) Fan,L.T.;Li,Y.;Wu,D.;Li,Z.R.;Sun,C.C.Aust.J.Chem.2012,65,138.doi:10.1071/CH11334
(32) Oudar,J.L.;Chemla,D.J.Chem.Phys.1977,66,2664.doi:10.1063/1.434213
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
电力科技与环保(2022年3期)2022-07-15
浙江化工(2022年4期)2022-05-07
光谱学与光谱分析(2020年2期)2020-02-25
山东青年(2018年2期)2018-06-23
国外医药(抗生素分册)(2016年4期)2016-07-12
北方文学·下旬(2016年6期)2016-05-14
信息记录材料(2016年4期)2016-03-11
中学化学(2015年5期)2015-07-13
中学化学(2015年5期)2015-07-13
