
超高效液相色谱-串联质谱法测定腐竹中乌洛托品的含量
2014-09-20 12:44,,,
食品工业科技 2014年17期
, , ,
(1.北京市产品质量监督检验院,北京 101300;2. 北京市食品安全监控和风险评估中心,北京 100041)
超高效液相色谱-串联质谱法测定腐竹中乌洛托品的含量
吴颖1,张慧1,崔芳1,姜洁2
(1.北京市产品质量监督检验院,北京 101300;2. 北京市食品安全监控和风险评估中心,北京 100041)
建立用超高效液相色谱-串联质谱法(UPLC-MS/MS)测定腐竹中乌洛托品含量的方法。用乙腈提取腐竹中的乌洛托品,经固相萃取柱PXA净化,用UPLC-MS/MS分析,采用多反应监测模式,外标法定量。采用ACQUITY UPLC BEH HILIC色谱柱,以乙酸铵和乙腈为流动相,进行梯度洗脱。结果表明:该方法标准曲线良好,线性范围为1~20μg/L,最低检出限为1μg/L,三个水平添加浓度的平均回收率范围为81.67%~101.67%。本方法操作简单快捷,方法准确度和稳定性好。
超高效液相色谱-串联质谱法,腐竹,乌洛托品,检测
腐竹是一种人们喜爱的传统豆制品,但是其保存周期很短,为了使腐竹外表光鲜、保质期长,一些不法厂家在生产的过程中常非法添加硼砂和吊白块,经过多年的专项治理整顿,已经鲜少见到,但是他们的添加手法和技术也日趋先进和隐蔽,目前监管部门已经发现乌洛托品已经作为吊白块的替代品添加到腐竹中。
乌洛托品(urotropine),又名六亚甲基四胺(hexamethylenetetramine),本身是属于低毒类,常应用于医药和工业上。作为非法添加物,它主要起到防腐,增白,改善产品外观的作用,其原理在于乌洛托品在酸性条件下可以分解产生甲醛,对人体的肾、肝、中枢神经、免疫功能、消化系统等均有损害,严禁用于食品或食品加工过程中。目前,我国还没有检测腐竹中乌洛托品的标准,仅有行业标准SN/T 2226-2008应用于鸡肉、鸡肝脏、鸡肾脏和猪肉中乌洛托品的检测[1]。相关文献报道应用于腐竹中的检测目前已有少数报道,有激光拉曼光谱法[2]、气相色谱法[3-4]、液相色谱法[5]、和液质联用法等方法[6-7],因此开展相关检测工作很有意义。本文建立了腐竹的超高效液相色谱及串联质谱方法,此方法的开发,可大大减少样品分析时间。
1 材料与方法
1.1材料与仪器
乌洛托品标准品 购于sigma公司,纯度≥99%。100μg/mL标准品储备液由乙腈溶解,并置于4℃冰箱中密封保存。色谱纯甲醇、乙腈和乙酸铵 购于Dikma公司。实验用水为超纯水。固相萃取柱 购于DIKMA公司的ProElut PXA,规格为150mg/6mL。
Waters ACQULITY UPLC超高效液相色谱仪;Waters Quattro Premier XE质谱仪(Waters,USA);高速冷冻离心机(SIGMA,德国);均质机;氮吹仪;ACQULITY UPLC BEH HILIC色谱柱2.1×50mm,1.7μm。
1.2色谱分析条件
液相色谱操作条件:流速:0.3mL/min;柱温:40℃;进样体积:10μL;流动相:A为10mmol/L 乙酸铵溶液,B为乙腈;梯度洗脱条件如表1。
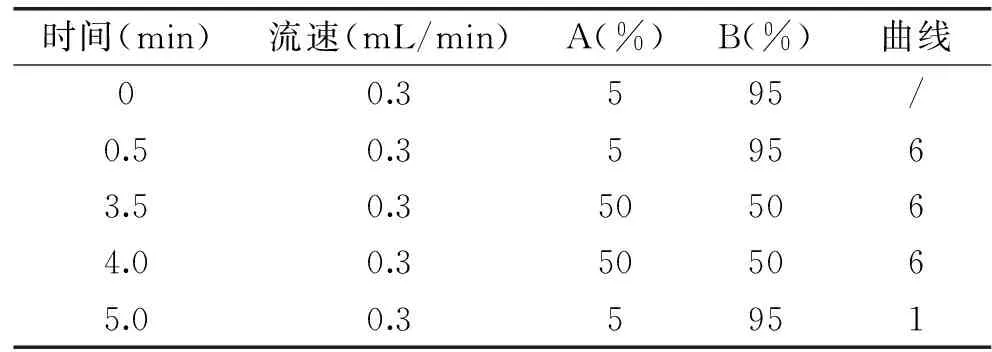
表1 液相梯度条件Table 1 Condition of gradient elution
质谱离子源选用电喷雾离子源(ESI);扫描模式:正离子扫描(ESI+);检测方式:多反应检测;干燥气:氮气;离子源温度:110℃,脱溶剂温度:350℃,脱溶剂气:600L/h,锥孔气:50L/h。质谱分析条件见表2。
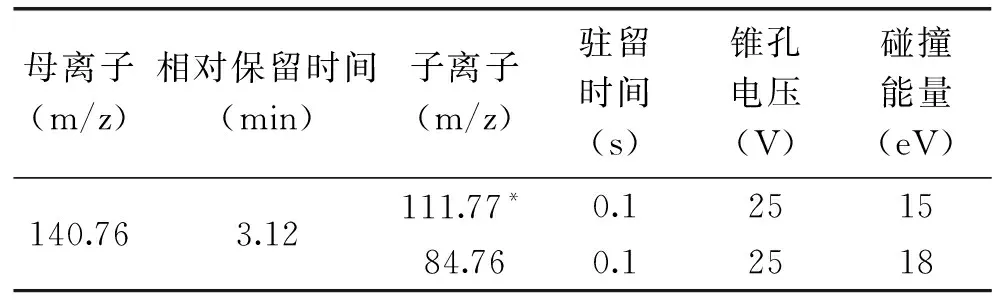
表2 质谱分析参数Table 2 Parameter of mass spectra
注:*标注的子离子为定量离子。
1.3标准溶液的配制和曲线绘制
精密称取乌洛托品1mg溶解于10mL乙腈,配制成浓度为100μg/mL的标准品储备液,将浓度为100μg/mL的标准品储备液稀释100倍,配成浓度为1μg/mL的标准使用液,再配制成工作液,浓度梯度为1,2,5,10,20μg/L,绘制标准曲线。
1.4样品前处理
1.4.1 提取 称取1.0g腐竹(精确至0.01g)(已粉碎均匀)和1.0g无水硫酸钠于聚四氟乙烯离心管中,加入10mL乙腈溶液,涡旋混合2min,超声10min,10000r/min离心10min,取上清液待净化。
1.4.2 净化 将PXA小柱用5mL乙腈活化,将1.4.1中上清液加入柱中,收集流出液(1),再加入1mL乙腈,收集流出液(2),合并流出液,用N2吹干,用90∶10的乙腈∶水溶解定容至1mL,涡旋混匀。过 0.22μm 滤膜,待上机使用。
2 结果与分析
2.1样品前处理条件的选择
腐竹样品采用乙腈提取,是由于乌洛托品在乙腈中溶解度高,可以保证较好的提取效果。净化过程选用了保留干扰物模式,PXA属于强阴离子交换小柱,可以很好的吸附脂肪酸类和氨基酸类的杂质,上样后,乌洛托品在小柱上面没有保留,随之流出。本实验通过氮吹的方式,进行富集浓缩,再用乙腈∶水=90∶10定容,较好的排除了基质的干扰,操作简便快速,提高了检测灵敏度和准确性,具备较好的重现性和稳定性。
2.2液相色谱-质谱条件的选择
BEH HILIC柱是一种可以保留一些强极性物质的色谱柱,这些物质在普通的C18的保留性差或者无保留,有分离效果不好、峰型拖尾等问题,因此本文没有采用C18柱,选用BEH HILIC色谱柱。本实验尝试了含有甲酸的水溶液-乙腈、水-乙腈和含有乙酸铵的水溶液-乙腈进行比较,发现含有乙酸铵的水溶液-乙腈的流动相分离的效果好,峰型对称。这也是BEH HILIC色谱柱的特性,可以结合高比例有机相/低比例水相组成的流动相来保留目标物质,同时这样的流动相组成也有利于提高电喷雾离子化质谱(ESI-MS)的灵敏度。在ESI+电离方式下,进行了仪器的毛细管电压、锥孔电压、碰撞能量等参数的优化选择。如图1和图2分别为乌洛托品的总离子流图和多反应监测色谱图,其中图2分别为是标样母离子打碎后的选定的两个碎片离子的图,定量离子图和定性离子图。在样品中,目标峰可以有很好的响应,和杂峰分离良好。
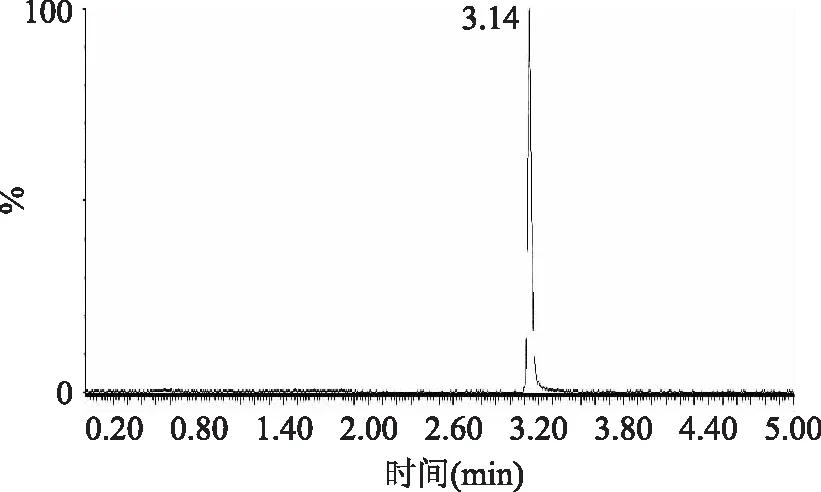
图1 总离子流图Fig.1 Total ionic chromatographic

图2 多反应监测色谱图Fig.2 Multiple reaction monitoring chromatogram
2.3方法线性范围、测定低限及精密度
将工作液分别进样10μL,得到回归方程(Y为峰面积,X为浓度,μg/L)Y=174.564×X+0.775512,线性相关系数R为0.998,在1~20μg/L浓度范围内成良好的线性。标准曲线如图3所示。
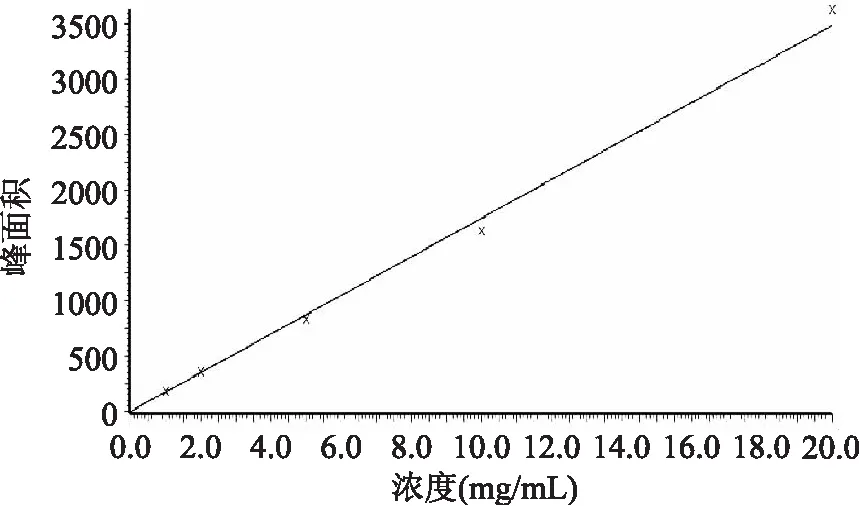
图3 标准曲线Fig.3 Calibration curve
在腐竹空白样品中添加低、中、高三个含量的乌洛托品,按照方法操作步骤进行回收率实验,同时每个添加浓度平行重复6次,测定精密度,以S/N≥10计的测定低限和标准偏差结果见表3所示。由表3可以看出,加标回收率在81.67%~101.67%,其多次测定的RSD在1.84%~3.99%,方法的准确度和精密度满足分析要求。腐竹空白样品上添加浓度为2μg/L的色谱图见图4。
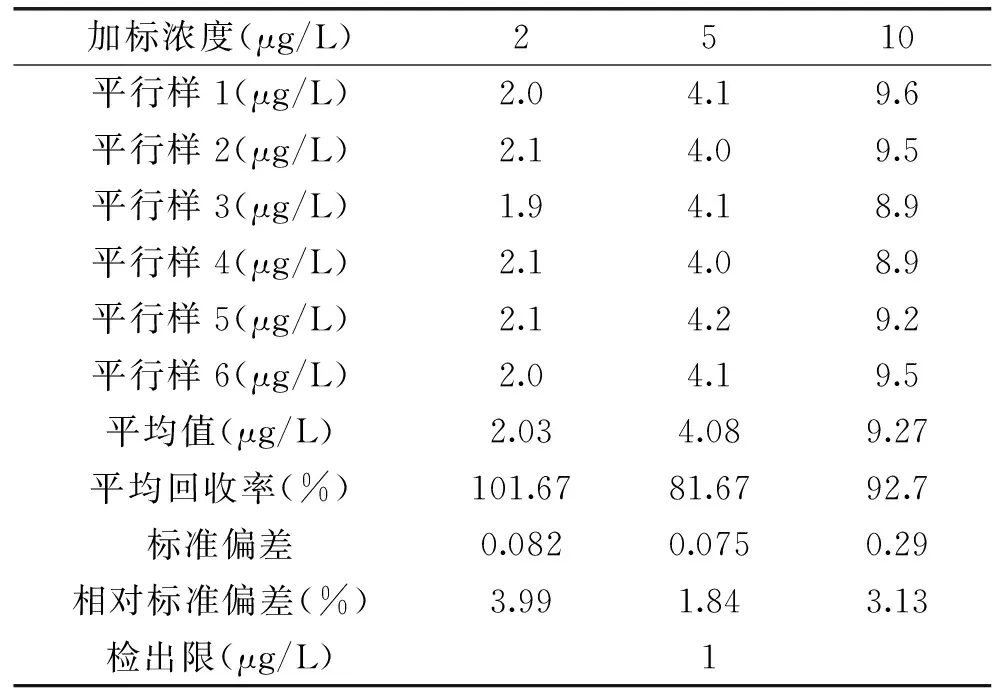
表3 回收率与相对标准偏差(n=6)Table 3 Recoveries and relative standard deviations(n=6)

图4 添加乌洛托品标准品(2μg/L)的多反应监测色谱图Fig.4 MRM chromatogram of a sample spiked with urotropine at 2μg/L
3 结论
本研究建立了固相萃取净化、液质联用检测腐竹中乌洛托品含量的方法,实验结果表明,该方法操作简单快捷,准确度和稳定性好。应用该法检测了市场和商超6个不同品牌的腐竹产品,均未发现有乌洛托品的残留。另外本实验仅探讨了腐竹中的乌洛托品的含量,其余的豆制品的分析尚需进一步探讨。
[1]SN/T 2226-2008 进出口动物源性食品中乌洛托品残留量的检测方法液相色谱-质谱/质谱法[S].
[2]张涛,李战海,李炎,等.非法食品添加剂乌洛托品的拉曼光谱及DFT分析方法[J].检验检疫学刊,2013,(2):36-37.
[3]李薇,陈天科,徐辉,等.乌洛托品的气相色谱分析[J].分析仪器,2007,(3):29-31.
[4]黄国春.气相色谱法测定腐竹中乌洛托品含量的研究[J].广西轻工业,2008,(6):26-27.
[5]李莉,张钢平.高效液相色谱法测定乌洛托品片的含量[J].中国医院药学杂志,2009,(4):335-336.
[6]洗燕萍,陈立伟,罗东辉,等.UPLC-MS/MS测定腐竹和米粉中的乌洛托品[J].江南大学学报(自然科学版),2012,(11):78-82.
[7]陆春良,刘娟,刘向农.亲水作用液相色谱电喷雾串联质谱法测定米线中乌洛托品残留[J].扬州大学学报:农业与生命科学版,2013,(2):82-86.
Determination of Urotropine in dried beancurd sticks by ultra performance liquid chromatography-tandem mass spectrometry
WUYing1,ZHANGHui1,CUIFang1,JIANGJie2
(1. Beijing Products Quality Supervision and Inspection Institute;Beijing 101300,China;2. Beijing Municipal Center for Food Safety Monitoring and Risk Assessment;Beijing 100041,China;)
A method for the determination of urotropine in dried beancurd sticks was established by UPLC-MS/MS.The sample was extracted with acetonitrile as extraction solvents,then further purified by PXA solid phase extraction column. The analyte was determined by multi-reaction monitoring(MRM)mode was employed for the quantitative determination,and quantified by the external standard curve.The separation was performed on a UPLC ACQUITY BEH HILIC column by gradient elution with a system of water(containing 10mmol/L ammonium acetate buffer)-acetonitrile as mobile phase.Results showed that the calibration curve had good linearity.The developed method exhibited good linearity over the range from 1 to 20μg/L.The minimum detection limit was 1μg/L. The average recovery rate for urotropine was 81.67%~101.67%.The method is simple,accurate and suitable for the identification and quantification.
Ultra performance liquid chromatography-tandem mass spectrometry;dried beancurd sticks;urotropine;determination
2014-01-24 *通讯联系人
吴颖(1965-),女,高级工程师,研究方向:生物化工。
TS
A
1002-0306(2014)17-0000-00
10.13386/j.issn1002-0306.2014.17.001
猜你喜欢
中国调味品(2023年1期)2023-01-13
煤化工(2022年3期)2022-07-08
食品安全导刊(2021年21期)2021-08-30
保健与生活(2021年15期)2021-08-16
色谱(2021年7期)2021-06-07
理化检验-化学分册(2020年12期)2021-01-26
农家之友(2019年3期)2019-01-16
食品界(2018年8期)2018-09-03
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
