
CL-20结晶形貌的预测
2014-09-11 02:40:26闻利群王美妮
山西化工 2014年2期
文 国, 闻利群, 王美妮, 王 宁
(中北大学化工与环境学院,山西 太原 030051)
引 言
六硝基六氮杂异伍兹烷(CL-20)是一种著名的高能钝感炸药[1],其爆轰性能和感度大小在一定程度上受到晶体形态的影响。CL-20分子具有偶极子的特性,其结构的共价键具有方向性和饱和性,生长基元在结晶方位和晶体各个面族上叠合的稳定性与结晶型态有关[2],且CL-20晶体的形态特征除了与其内部结构有关之外,还与生长时的物理、化学条件密切相关[3],如溶剂、过饱和度、杂质等,因此研究CL-20晶体的生长习性需要把晶体内部结构同生长条件密切结合起来,这是研究晶体生长机理的一个重要途径。模拟晶体生长或预测晶习的基本物理模型有 Bravais提出的 BFDH 模型[4]、Hartman和Perdok提出的PBC模型、Bennema等提出的附着能(attachment energy,简称 AE)模型[5]。其中,基于AE模型预测晶习被证实是较为精确的[1-4,6-7]。该模型认为,晶面附着能绝对值越大,生长速率越快,重要性越低,该模型被广泛用于研究晶体生长的热动力学、晶面生长机理、多晶生长的界面结构等。Jianxin chen等[4]分别用BFDH模型和AE模型对氢化可的松在甲醇中的晶貌进行预测,并与实验对比。结果表明,AE模型的准确性较高。A.V.D.Heijden等[1]用AE模型对RDX结晶形态进行了预测,发现(111)、(200)和(020)等面为重要生长面。郁卫飞等[6]进行了以水和NMP为混合溶剂的CL-20重结晶实验,发现在10℃/min的降温速率下晶粒较规整。薛其彬等[7]进行了CL-20挥发结晶实验,并通过XRD实验分析出分型结构的晶体可能是某些面择优取向的生长结果。
但对于CL-20晶貌形成的内在机理,微观层次上溶剂分子对CL-20晶习的作用机制和改变情况,尚未有人做过深入细致的研究。本研究对AE模型预测出的CL-20在真空中的晶貌从键链角度分析其形成机理,根据溶剂与各晶面的交互作用,用修正附着能的方法对CL-20在水和甲醇中形貌进行了预测,并分析了其成因。
1 AE晶体生长模型
AE模型是在PBC模型的基础上发展起来的。PBC模型假设晶体表面成键所需时间与键合能成反比,晶面生长速度正比于键合能,晶体结构由PBC组成,且晶体生长最快的方向是化学键最强的方向[9]。而AE模型在此基础上定义了晶片能Ea为生长出一层厚度为dhkl的晶片释放出的能量,附着能Eatt为晶片附着在晶体表面(hkl)所释放出的能量,两者之和为晶体的晶格能Elatt[8],见式(1)。

对于有机分子,晶格能可通过非成键原子间作用力函数[2]和分子间作用综合计算求得;对于晶片能和附着能,可由中心分子与晶片内、外的所有分子总作用力计算得出。
Berkovitch-Yellin[9]对前述方法加以改进,指出各晶面的相对生长速率(Rij)正比于附着能Eatt,见式(2)。

最低附着能的晶面生长得最慢,所以具有最大的形态学特征,从而决定了晶体的微观形貌。该方法基于晶体对称性、分子间键链特性和晶面附着能来计算、推演出有机分子在真空和溶剂中的附着能、晶貌特征。
2 晶面-溶剂的吸附能计算
2.1 晶体与溶剂分子模型的构建
计算所需的CL-20晶胞参数均在CCDC数据库查得,具有α和β2种晶型,均属于单斜晶系。因后者密度较高,在起爆药内应用较多,故本研究对象为β型CL-20晶体。
根据β型CL-20晶体中原子分数坐标和对称性信息在Material studio 6.0中建立原胞,并在Discover模块下进行5 000步的几何优化。因分子动力学模拟(MD)过程需要较大的数据采样空间,故用优化后的原胞建立4×4×4的超晶胞。为进一步消除CL-20超胞中不合理的能量和构象,用Forceit模块下Anneal过程对其进行300℃~500℃升、降温退火循环处理,此过程步数为10 000步,采用Compass力 场[10]、NPV 系 综、Andersen 控 温 方法[11],各原子起始速度按 Maxwell分布取样,Velocity Verlet[12]算法进行求解,范德华和静电作用,分别用 Atom based[13]和 Ewald[14]方法,原子截断半径取1.25nm,缓冲宽度取0.001。此过程可对充分豫弛超胞,为以后的MD模拟提供合理、平衡的几何构象。
水和甲醇分子的模型由Sketch建立后,也采用前述方法进行几何构型和能量的豫弛。
2.2 吸附模型的构建与优化
溶剂分子对CL-20晶习的影响是通过与其晶面的吸附作用实现的,故需先建立水、甲醇与CL-20晶面的吸附模型,再后求解其吸附能。
对前面退火所得到的CL-20超胞最低势能构象的100、001和010面进行切割(之前,用Crystal-Graph基于单体键链能量和方向对CL-20晶体生长面做过预测,得知100、-100、001、00-1、010和0-10晶面是易保留下来的重要生长面。因CL-20所属晶系的高度对称性,100和-100、001和00-1、010和0-10互为同一面且方向平行。为减小计算量,仅取100、001和010面研究),切割厚度为2个分子层。之后,对此二维结构加2nm的真空层(vacuum),由此形成了具有吸附面的层晶(slab)。分别往各个吸附面加入水和甲醇分子,用文献[7]中的方法调整合理的构型,以形成6个初始吸附模型,再对每种模型进行几何优化,方法与前述相同。经优化后的水、甲醇与100面吸附模型见图1、图2(虚线为氢键)。其中,图1中的吸附模型与文献[15]中的CL-20和水全优化构型相符。

图1 水分子与100面的吸附模型
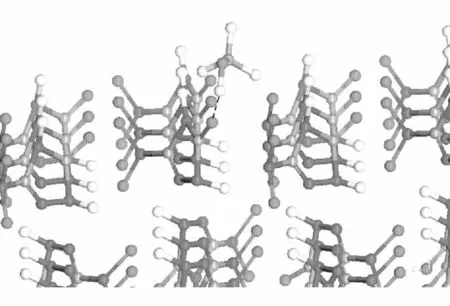
图2 甲醇分子与100面的吸附模型
2.3 吸附过程的动力学模拟
用Forceit模块下的Dynamic过程对前面优化后的6种吸附模型进行NPT系综下的MD模拟,总时间为150ps,步长为1fs,其他参数与退火过程相同。MD过程结束后,用各模型能量稳定后的构型(后50帧)分别对整个体系、溶剂分子和晶面的能量求均值,之后用吸附能计算公式[4,16-17](3)进行吸附能求解,计算结果见第29页表1。

式中:Etotal为吸附体系的总能量,kcal/mol;Esolv为溶剂分子单点能,kcal/mol;Esury为晶面的表面能,kcal/mol。

表1 溶剂分子与晶面的吸附能
3 CL-20晶习的预测
预测CL-20真空中晶习的主要目的是从晶体的周期性键链(即分子间作用)和自身附着能的角度分析自由状态下晶貌生成的内部因素,并据此用溶剂分子与晶面吸附和对附着能改变的方法预测和研究CL-20在水、甲醇中的晶习。
3.1 真空中的晶习
采用Crystal Graph法计算优化后CL-20原胞晶面间的交互作用(见图3)和各晶面上CL-20分子的附着能值(见表2),根据Rij∝Dij∝Eatt[6]可求得各面的相对生长速率和生长面到晶体中心的距离Dij,再通过Wulff图对上述信息进行分析,推导出真空环境下晶体的生长形态(见图3)。重要面的生长情况见表2。

图3 真空中NTO晶貌和分子间作用力

表2 真空中CL-20重要晶面生长状况
CL-20分子在010方向形成了一种偶极生长基元,(—NO2)基团方向电子云密度大,为负极面,与三唑环上的正电荷中心(距C原子较近)有很强的分子间力。010晶面与晶体最强键链方向夹角小,受此极化作用影响,CL-20分子在010面叠合效率较高,故010面的生长速率较100和001面快、晶面面积小,相对于其他快速生长、或已消失的面仍顽强显露。
001面和100面几乎垂直于CL-20分子的正-负极面。100晶面方向的分子附着主要靠羰基与三唑基团间微弱的亲和作用;001晶面方向与三唑环共轭平面夹角小,分子附着主要靠共轭体系间的范德华力作用。由于偏离极性生长方向,CL-20分子间叠合能小,晶面生长缓慢,故100和001面在结晶过程中能保留下来且成为重要的生长面。
由于Rij∝Eatt,故真空中各晶面生长速率010>>100≈001,其形貌为长四棱柱。
3.2 溶剂中的晶习
3.1 中的模拟结果未考虑溶剂及其他因素对晶体形貌的影响,所以需要对其进行进一步修正。考虑到溶剂对不同晶面的作用不同,CL-20分子附着能计算公式为式(4)[4,16]:

式中:Em是修正后CL-20分子的附着能,kcal/mol;Es是晶面对溶剂分子的吸附能,kcal/mol;Eatt是真空中CL-20分子的附着能,kcal/mol。计算结果见表3。由表3可知,溶剂分子对晶习的影响通过其与晶面的吸附来影响CL-20分子在各面的叠合能量与效率,溶剂与面相互能愈大,面生长愈慢。反之亦然。

表3 真空、水和甲醇中CL-20主要晶面附着能
溶剂的吸附改变了CL-20各晶面间的交互作用。根据水、甲醇与不同晶面结合强度(吸附能)的不同,修正各晶面的附着能值,为各晶面指定相对生长速率和中心到面的距离,再通过Wulff图对以上信息进行分析,推导出晶体形态,并与文献中CL-20在水和甲醇中的结晶型态电镜扫描图对比,见图4和图5。

图4 水中CL-20晶貌的预测和实际图[1]

图5 甲醇中CL-20晶貌的预测和实际图[1]
综合表1~表3分析可得,水和甲醇均会通过与晶面的吸附作用而降低CL-20分子的附着能。据文献[2]显示,溶剂与有机分子的相互作用中,除范德华力和静电力外,氢键的贡献尤为显著。
水分子可与010晶面上分布密集的硝基氧形成较强的氢键,其吸附作用最强;水分子中的氢、氧原子能与CL-20分子上的9位氧原子、11位氢原子[16]间形成2个氢键,且方向垂直于100面,该方向的吸附强度次之;三唑共轭平面与001面的夹角小,且无垂直于该面方向的氢原子显露,故001面与水吸附能最低。溶剂与面吸附能愈大,CL-20分子附着能越小,且Rij∝Eatt,但010面的附着能本身很大,溶剂的强抑制作用并不能使其显著降低,所以水中各面生长速率为010>001>100。从图3可见,其结晶型态接近扁平的六面体。
甲醇分子同各面的相互作用机理类似于水。另外,甲基推电子基团使羟基周围电子云密度高,分子偶极距大[1,4],与各面形成的氢键作用较强且键能差距较小,但在100面所成的氢键无水分子与该面所成氢键的特殊性,故与100面的吸附强度与水相当。甲醇中各面的生长速率为010>100≈001,从图4可见其结晶型态为四棱柱。
用修正后的AE模型所预测的晶习与扫描电镜显示的实际晶习能很好地相匹配,说明预测是成功的。
4 结论
1)利用AE模型和修正附着能的方法可对CL-20晶体在真空、水和甲醇环境中的晶习进行预测,其结果和实际能较好地吻合。
2)真空中CL-20晶体各面的生长速率受垂直于面方向的分子间力影响,生长速率依次为010>100≈001,其形貌为长四棱柱。
3)受水和甲醇抑制作用的影响,在水中晶面生长速率依次为010>100>001,晶体外观为扁平的六面体;甲醇中为010>100≈001,晶体外观为四棱柱,较真空中的稍短。
[1]Heijden A V D,Horst J.Energetic materials particle processing and characterization[M].Weinheim:WILEY-VCH Verlag GmbH & Co.KGaA,2005:84-139.
[2]仲维卓,华素坤.晶体生长形态学[M].北京:科学出版社,1999:390-400.
[3]薛其彬.表面诱导NTO晶体生长研究[D].绵阳:西南科技大学,2009.
[4]Chen Jianxin,Wang Jingkang,Zhang Ying,et al.Crystal growth,structure and morphology of hydrocortisone methanol solvate[J].Journal of Crystal Growth,2004,161(3):265-273.
[5]Hartman P,Bennema P.The attachment energy as a habit controlling factor:theoretical considerations[J].Cryst Growth,1980,49:145.
[6]郁卫飞,聂福德.类球形NTO的制备及表征[J].含能材料,2005(13):4-6.
[7]薛其彬,黄辉.NTO晶体生长:从分型结构到立方结构[J].光谱学与光谱分析,2009(17):381-384.
[8]陈建新,王静康.氢化可的松形貌预测[J].天津大学学报,2006,39:3-6.
[9]Berkovitch-Yellin Z.Toward an ab initio derivation of crystal morphology [J].J Am Chem Soc,1985,107(26):8239-8253.
[10]Sun H.Compass:an ab initio force field optimized for condensed phase applications overview with details on alkane and benzene compounds[J].J Chem Phys B,1998,102(38):7338-7364.
[11]Andersen H C.Molecular dynamics simulations at constant pressure and/or temperature[J].J Chem Phys,1980,72(4):2384-2393.
[12]Allen M P,Tildesley D J.Computer simulation of liquids[M].Oxford:Clarendon Press,1987:78-80.
[13]Karasawa N,Goddard W.Force fields,structures,and properties of poly crystals[J].Macromolecules,1992,25(26):7268-7280.
[14]Ewald P P.Evaluation of optical and electrostatic lattice potentials[J].Annalen der Physik,1921,369(3):255.
[15]方国勇,徐丽娜.3-硝基-1,2,4-三唑-5-酮与 NH3及H2O分子间相互作用的理论研究[J].化学学报,2005(63):1056.
[16]王相元.太安重结晶球形化工艺研究[D].太原:中北大学,2009.
[17]段晓惠,卫春雪,衣重华,等.HMX晶体形貌预测[J].含能材料,2009,17(6):655-658.
猜你喜欢
火炸药学报(2022年5期)2022-11-04 02:30:48
人工晶体学报(2021年10期)2021-11-26 02:39:18
上海理工大学学报(2021年3期)2021-07-20 08:04:10
人工晶体学报(2021年6期)2021-07-12 07:59:58
中国材料进展(2019年10期)2019-12-07 05:32:54
人工晶体学报(2019年8期)2019-09-18 02:53:02
物理实验(2019年7期)2019-08-06 05:35:56
航空材料学报(2019年2期)2019-04-15 01:04:08
物理学报(2018年22期)2018-12-18 05:58:28
机械制造与自动化(2014年1期)2014-03-01 04:21:57
