
笔管草药材质量标准研究
2014-08-29 06:28刘大伟赖佐发廖宝源
江西中医药大学学报 2014年6期
★ 刘大伟 赖佐发 廖宝源
(赣州市食品药品检验所 江西 赣州 341000)
笔管草为木贼科植物笔管草EquisetumdebileRoxb.的干燥的地上部分。全年可采,割取地上部分,除去杂质,晒干。用于急性结膜炎,急性黄疸型肝炎,痢疾,尿道炎,荨麻疹。微苦、甘,平。归肝经。清热利湿,明目退翳。分布于江西(赣州)、广东、福建(泉州、厦门、龙岩、三明、南平)等省区,生于山谷溪边潮湿地,为江西省赣南地区民间常用药[5]。本品收载于《卫生部药品标准》中药成方制剂第九册(1994年版),原标准只有简单的性状、显微鉴别和化学反应,未对药效成分进行定性、定量检测,难以保障其药材及其成方制剂的质量和临床用药的有效性和安全性。[2]因此,本文在笔管草原标准基础上,建立了水分、总灰分、酸不溶性灰分、浸出物、山柰素的薄层鉴别和含量测定方法,为笔管草药材及其成方制剂的质量控制提供了依据。
1 仪器与试药
Agilent1260高效液相色谱仪(VWD检测器、四元低压混合泵、柱温箱、自动进样器、ChemStation色谱工作站);DM500摄影摄影显微镜(德国莱卡;USC302超声波清洗器(上海波龙电子设备有限公司);GZX-DH-40ⅹ45-BS电热恒温干燥箱(上海跃进医疗器械厂);SX2-5-12 C0149箱式电阻炉(上海实验仪器厂有限公司);梅特勒AB265-S型电子天平。
乙腈为色谱纯,水为重蒸馏水,其它试剂均为分析纯。山柰素对照品(中国药品生物制品检定所,批号:110861-200808,含量为95.9%)。10批笔管草药材为采自赣州市信丰小江(暂定批号为1 -10),经赣州市食品药品检验所刘志辉鉴定为木贼科植物笔管草EquisetumdebileRoxb.的干燥的地上部分。
2 方法与结果
2.1 植物来源[5]多年生草本,高18~100cm,硬且粗糙。根茎横走,黑或黑褐色。茎绿色,直立,直径2~15mm,中空,基部节上有2~5分枝,稀无分枝或仅有1小枝,有棱脊6~20条,棱脊上有1列小疣状突起或小横纹,沟中有气孔线1列。叶小,轮生,下部联合成鞘筒,疏松地包围茎节,鞘片背上无棱脊,鞘齿短三角形,黑色,有易脱落的膜质尖尾,孢子束穗生于分枝及主茎顶端,矩圆形,有小尖头,长0.5~2.5cm,无柄;孢子同型,圆球状,有长弹丝4,或十字形,平时紧绕在孢子外面,遇水即弹开。孢子成熟8~10月。
2.2 药材性状 本品呈长杆状,长30~50cm,直径0.2~0.5cm,表面灰绿色或黄绿色,有6~20条棱背,节明显,节间长2~9cm,叶鞘基部和鞘齿黑棕色,鞘齿短三角形,有的可见膜质尖尾,每节轮生枝2~5条。体轻,质脆,易折断,手捻之有粗糙感,断面中空,内表面白色膜质。气微,味淡微甘。
2.3 薄层色谱鉴别
2.3.1 供试品溶液的制备 取本品粉末1g,加75%甲醇25mL,盐酸1mL,加热水解1 h,滤过,滤液蒸干,残渣加水10mL溶解,用乙酸乙酯提取2次,每次10mL,合并乙酸乙酯液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。[1]
2.3.2 对照品溶液的制备 另取山柰素对照品,加甲醇制成每1mL含1mg的溶液,作为对照品溶液。
2.3.3 薄层色谱的展开 照薄层色谱法(《中国药典》2010年版一部附录Ⅵ B)试验,吸取上述两种溶液各1μL,分别点于同一硅胶G薄层板上,采用双槽展开缸,以环已烷-乙酸乙酯-甲酸(8∶4∶0.4)为展开剂,上行展开,展距8~15 cm。
2.3.4 显色 5%三氯化铝乙醇溶液,立即置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。

1. 山奈素;2~11. 笔管草药材
2.3.5 样品检识 按上述条件对10批土黄连药材进行薄层色谱鉴别,结果各斑点分离良好,显色清晰,且各批药材薄层鉴别结果一致性较好。见图1。
2.4 检查
2.4.1 水分 按照《中国药典》2010年版一部附录Ⅸ H第一法测定[8]。见表1。
2.4.2 总灰分 按照《中国药典》2010年版一部附录Ⅸ K测定。见表1。
2.4.3 酸不溶性灰分 按照《中国药典》2010年版一部附录Ⅸ K测定。见表1。
2.5 浸出物 用水作溶剂作溶剂,水溶性浸出物测定法(《中国药典》2010年版一部附录Ⅹ A)项下的热浸法测定。见表1。

表1 笔管草水分、总灰分、酸不溶性灰分、浸出物、含量测定结果


图2 对照品溶液(A)和供试品溶液(B)的HPLC色谱图
2.6 含量测定 笔管草主要含山柰素,因此本文采用HPLC法建立了笔管草药材中山柰素含量测定方法。
2.6.1 色谱条件 色谱柱:Diamonsil(钻石)C18柱(4.6mm×250mm,5μm);流动相:乙腈-0.4%磷酸溶液(50∶50);检测波长:365nm;进样量:10μL;流速:1.0mL/min;柱温:35℃。理论板数按山奈素峰计算应不低于3000。见图2。
2.6.2 对照品溶液的制备 取山奈素对照品适量,精密称定,加75%甲醇制成每1mL含20μg的溶液,即得。
2.6.3 供试品溶液的制备 取本品粉末(过三号筛)约0.75g,精密称定,置具塞锥形瓶中,精密加入75%甲醇50mL,密塞,称定重量,加热回流1 h,放冷,再称定重量,用75%甲醇补足减失的重量,摇匀,滤过,精密量取续滤液20mL,加盐酸5mL,置水浴中加热水解1 h,放冷,转移至50mL量瓶中,加75%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。[1]
2.6.4 线性关系考察 精密吸取1.0,2.5,5.0,7.5,10.0μg/mL的山奈素对照品溶液,分别进样10μL,测定。以进样量(μg)为横坐标,山奈素峰面积为纵坐标绘制标准曲线,回归方程为Y=4046.3X-18.885,r=0.9993。结果表明山奈素在0.00998~0.0998μg范围内线性关系良好。
2.6.5 精密度试验 取本品(批号20130116)0.75g,精密称定,按供试品溶液的制备方法制备,精密吸取10μL注入液相色谱仪,测定,重复6次,计算山奈素RSD=0.46%,表明精密度良好。
2.6.6 重复性试验 取本品(批号20130116)6份,每份0.75g,精密称定,按供试品溶液的制备方法制备,精密吸取10μL注入液相色谱仪,测定,记录山奈素峰面积并计算RSD=0.54%, 表明方法重复性良好。
2.6.7 稳定性试验 取本品(批号20130116)0.75g,精密称定,按供试品溶液的制备方法制备,分别于0,2,4,6,8,12,24h测定,记录山奈素峰面积并计算RSD=0.38%,结果表明供试品在24h内稳定。
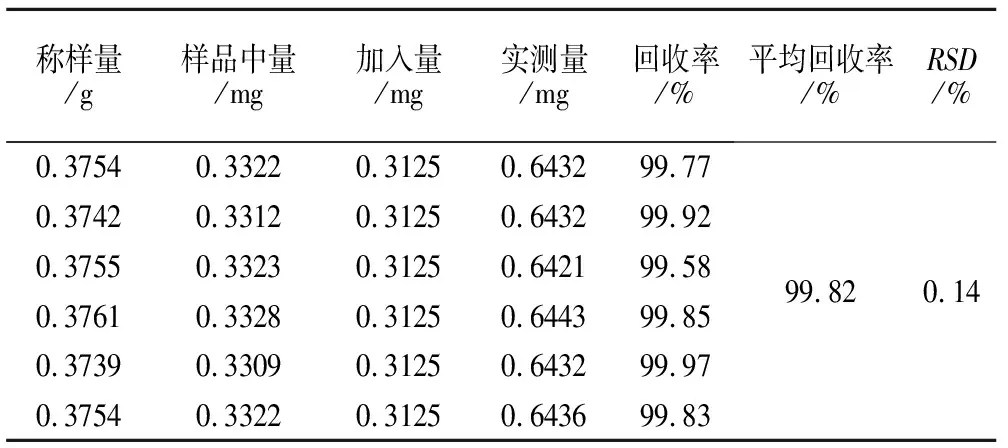
表2 山奈素加样回收率试验
2.6.8 加样回收率试验 取本品(批号20130116)6份,每份0.375g,精密称定,分别加入0.0125mg/mL的山奈素对照品溶液25mL,按供试品溶液的制备方法制备,精密吸取10μL注入液相色谱仪,测定,计算山奈素的含量及RSD。结果山奈素的平均回收率为99.82%,RSD=0.14%,表明该方法回收率良好。见表2。
2.6.9 药材含量测定 取本品细粉约0.75g,精密称定,按供试品溶液的制备方法制成供试品溶液,按上述色谱条件分别测定山柰素的含量,结果见表1。
3 讨论
3.1 薄层色谱鉴别 实验考察了供试品溶液的稳定性,每隔一定时间点样一次,在放置0,2,6,24 h内点样,结果表明:在2 h内,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。证明供试品溶液要临用新配。
3.2 浸出物测定考察 实验分别采取热浸法和冷浸法,热浸法中用水、50%乙醇、75%乙醇、乙醇分别做溶剂进行考察;冷浸法中用水、50%乙醇、75%乙醇、乙醇做溶剂进行考察,结果表明:水作溶剂热浸浸出率最高。故选取水作溶剂热浸。
3.3 灰分和酸不溶性灰分测定考察 针对本品灰分和酸不溶性灰分测定值较高,实验分别采取水洗和加10%硝酸铵溶液湿润后测定。实验所用样品均为干燥的地上部分,考虑到样品是否已洗净,如果样品带有灰尘和泥沙,均会影响最后的检测结果。所以,第一步采取水洗,净制样品,并延长炭化时间。水洗过程中,未发现有尘土和泥沙。样品的水洗前和水洗后的测定结果比较接近。考虑到考虑到样品炭化后有可能不易灰化,故采取加10%硝酸铵溶液湿润。具体操作为:当样品炭化后,放冷,加10%硝酸铵溶液3mL,使残渣湿润,置水浴上蒸干,再放进高温炉内进行炽灼,至坩埚内容物完全炭化。测定结果比较接近。因此,灰分和酸不溶性灰分测定值较高是样品成分原因。
3.4 含量测定
3.4.1 波长选择 取山柰素对照品适量,用75%甲醇制成适当的浓度,以75%甲醇为空白,在400~300nm波长范围内进行光谱扫描,结果在365nm波长处有稳定的吸收,同时参考《中国药典》中山柰素的检测波长,确定检测波长为365nm。
3.4.2 流动相的选择 参考《中国药典》中山柰素含量测定方法,采用乙腈-0.4%磷酸溶液(50∶50)为流动相体系,山柰素与其它组分分离完全,结果满意。
[1]国家药典委员会·中华人民共和国药典(一部)[S].北京:中医医药科技出版社,2010:285-286.
[2]中华人民共和国卫生部药典委员会.卫生部药品标准﹒中药成方制剂第九册[S].北京:中华人民共和国卫生部药典委员会出版,1996:180-181.
[3]北京市卫生局.北京中药材标准[M].北京:首都师范大学出版社,1998:211.
[4]福建省食品药品监督管理局.福建省中药材标准[M].福州:海风出版社,2006:211.
[5]中国科学院植物研究所.中国高等植物图鉴(第一册)[M].北京:科学出版社,1972:116.
猜你喜欢
选煤技术(2022年2期)2022-06-06
选煤技术(2022年2期)2022-06-06
人人健康(2021年14期)2021-08-06
选煤技术(2021年6期)2021-04-19
四川蚕业(2021年4期)2021-03-08
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
中国猪业(2016年1期)2016-01-28
中国中医药现代远程教育(2014年11期)2014-08-08
中国医药生物技术(2014年4期)2014-01-23
