
参花胶囊的HPLC指纹图谱研究及4种成分含量测定
2014-08-20 08:36肖会敏谢艳华王四旺第四军医大学药学院新药研究中心西安710032
西北药学杂志 2014年6期
肖会敏,何 悦,党 珍,谢艳华,王四旺(第四军医大学药学院新药研究中心,西安 710032)
参花胶囊由红花、丹参、菊花等中药组成,主要用于活血化瘀、行气止痛等。处方中主要活性成分有:丹参中的丹参酮类、丹酚酸类[1-3],川芎中的川芎嗪、阿魏酸等[4],红花中的红花黄色素、羟基红花黄色素A等[5-6],赤芍中的单萜及单萜苷类化合物、苷类等[7],菊花中的黄酮类化合物和绿原酸等[8],山楂中的黄酮类、黄烷及其聚合物类、三萜类和有机酸类等[9],甘草主要活性成分含有多种黄酮类化合物、三萜类化合物以及香豆素类、氨基酸、多种生物碱和多种有机酸等[10-11]。本处方采用醇提与水提等工艺制成。为表征本处方中的主要活性成分,作者采用高效液相色谱(HPLC)法对该制剂进行分析,参照文献方法[12]进行实验;同时,对参花胶囊中的4种成分即绿原酸、羟基红花黄色素A、迷迭香酸、丹酚酸B进行定量分析。
1 仪器与试药
1.1仪器 岛津高效液相色谱仪,型号Prominence UFLC,配LC-20AD双泵,SPD-M20A二极管阵列检测器,SIL-20ACHT自动进样器,CTO-20A柱温箱,LC/Labsoluion色谱工作站(日本岛津公司);BSA124S电子分析天平(德国赛多利斯公司);KQ-300DE数控超声波发生器(昆山超声仪器有限公司)。
1.2试药 乙腈、甲醇,色谱级(美国Fish公司);超纯水,由美国Millipore纯水仪制备(美国millipore公司);丹酚酸B、迷迭香酸、绿原酸、羟基红花黄色素A对照品均购自中国药品生物制品检定研究院;参花胶囊(批号20090908,20090918,20090928,20130601,20130602,20130603,20130604,20130605,20130606,20130607),第四军医大学药物研究所研制。
2 指纹图谱测定方法与结果
2.1色谱条件 色谱柱:Kromasil C18(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-5.0 g·L-1磷酸水溶液溶液(B),梯度洗脱:0~15 min,A 5%→21%;15~40 min,A 21%→40%;40~65 min,A40%→100%;65~70 min,A100%。记录时间为70 min 。体积流量:0.8 mL·min-1;柱温:30 ℃;检测波长:254 nm;进样量5 μL。
2.2供试品与参比物溶液的制备
2.2.1参比物溶液的制备 精密称取迷迭香酸对照品5.0 mg,置于50 mL量瓶中,加体积分数70%甲醇至刻度,摇匀,即得质量浓度为0.1 mg·mL-1的参比物溶液。
2.2.2供试品溶液的制备 取10粒本品内容物,混匀,称取0.5 g,精密称定,置于10 mL量瓶中,加70%甲醇适量,超声30 min,放冷,加体积分数70%甲醇至刻度,滤过(0.45 μm滤膜),取续滤液作为供试品溶液。
2.3方法学考察
2.3.1精密度实验 取供试品溶液(批号20090908),连续进样6次,分别对共有峰的相对保留时间和相对峰面积进行分析。结果,各共有峰的相对保留时间和相对峰面积RSD值分别小于0.5%和2.0%,表明仪器精密度良好。
2.3.2稳定性实验 取供试品溶液(批号20090908),分别于各时间点即0,2,4,8,12和24 h进样,分析共有峰的相对保留时间、相对峰面积。结果,各共有峰相对保留时间、相对峰面积RSD值分别小于1.0%和2.0%,表明供试品溶液在24 h内稳定性良好。
2.3.3重复性实验 取6份样品(批号20090908),分别按供试品溶液的制备与色谱条件进行分析,分别对共有峰的相对保留时间和相对峰面积进行分析。结果,各共有峰相对保留时间与相对峰面积RSD值分别小于0.5%和2.0%,表明重复性良好。
2.3.4指纹图谱的建立及相似度评价 取10批样品,分别按上述供试品溶液的制备方法制备样品溶液,进样,得色谱图(图1),峰17为迷迭香酸即参比物,按上述色谱条件进行分析,建立指纹图谱。10批样品共有色谱峰的相对保留时间、相对峰面积的RSD值均分别小于0.5%和2.0%,符合文献[10]要求,见图1和图2。运用文献[10]要求对各批样品的指纹图谱进行相似度分析,对选定39个共有色谱峰进行谱峰匹配,通过计算得出样品指纹图谱的共有模式,并以此共有模式为标准,进行整体相似度评价,结果相似度为0.966~0.998,表明这各批样品的化学成分一致性较好。

图1 参花胶囊HPLC指纹图谱

图2 10批参花胶囊的HPLC指纹图谱
3 含量测定方法与结果
3.1色谱条件 检测波长为320 nm,其余同2.1项。
3.2对照品溶液的配制 分别精密称取绿原酸对照品2.2 mg、迷迭香酸3.8 mg、羟基红花色素A12.3 mg、丹酚酸B17.1 mg,置于10 mL量瓶中,加体积分数70%甲醇至刻度,作为储备液。
3.3供试品溶液的制备 同2.2.2项。
3.4线性关系 分别精密量取储备液0.031,0.062,0.125,0.250和0.500 mL对照品溶液,分别置于1 mL量瓶中,并加体积分数70%甲醇稀释至刻度,用0.45 μm微孔滤膜滤过,进样。以各对照品的质量浓度(x,μg·mL-1)为横坐标、峰面积值(y)为纵坐标,通过计算,得线性方程,结果见表1,表明各对照品在相应质量浓度范围内线性关系良好。
3.5精密度实验 精密吸取上述线性对照品溶液(0.125 mL储备液加体积分数70%甲醇定容至1 mL),重复进样5次,测得绿原酸、羟基红花黄色素A、迷迭香酸、丹酚酸B的相对标准差(RSD)分别为1.05%,1.07%,1.25%,1.09%和1.36%,表明仪器具有良好的精密度。
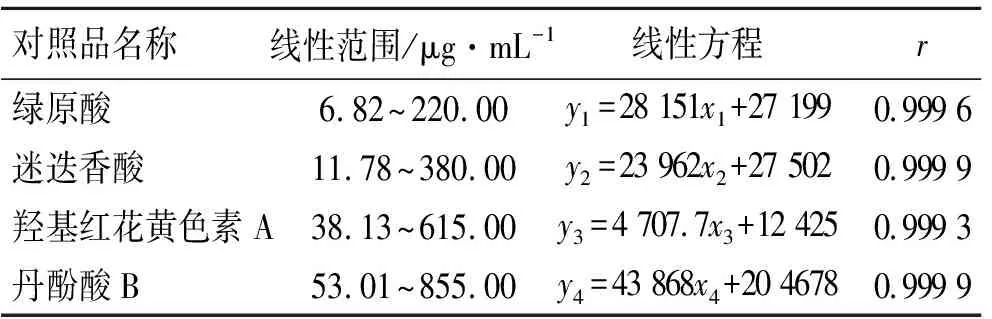
表1 对照品的线性方程及其质量浓度范围
3.6重复性实验 精密称取5份样品(批号20090908),分别按2.2.3项下方法制备供试品溶液,按3.3项下色谱条件进样测定。结果,绿原酸、羟基红花黄色素A、迷迭香酸、丹酚酸B平均含量分别为0.55,3.20,0.99和8.50 mg·g-1,RSD值分别为1.33%,1.05%,1.15%和1.42%,表明该方法具有良好重复性。
3.7稳定性实验 精密吸取对照品溶液(0.125 mL储备液,加体积分数70%甲醇定容至1 mL),在第0,2,4,8,12和24 h进样。结果,绿原酸、羟基红花黄色素A、迷迭香酸、丹酚酸B的峰面积的RSD值分别为1.08%,1.14%,1.19%和1.33%,表明该溶液在24 h内稳定。
3.8加样回收率实验 取参花胶囊(批号20090908,其中绿原酸、羟基红花黄色素A、迷迭香酸、丹酚酸B分别为0.55,3.20,0.99和8.50 mg·g-1)10粒内容物,混匀,称取粉末0.5 g,精密称定,分别添加混合对照品溶液(分别精密称取绿原酸对照品1.0 mg、迷迭香2.5 mg、羟基红花色素A 8.0 mg,丹酚酸B 8.4 mg,置于5 mL量瓶中,加70%甲醇至刻度)0.25 mL或0.50 mL或1.00 mL,按照2.2.3项下方法制备供试品溶液,共制备6份样品,按照3.1项下色谱条件进样测定。结果见表2,表明该方法回收率良好。

表2 加样回收率测定结果

表2(续) 加样回收率测定结果
3.9含量测定 精密称取10批样品,分别按照2.2.2项下方法制备供试品,按照3.1项下色谱条件测定。各批样品中绿原酸、羟基红花黄色素A、迷迭香酸 、丹酚酸B的平均含量分别为0.54,3.22,0.99和8.52 mg·g-1,RSD分别为1.37%,0.77%,1.40%和0.44%。
4 分析与讨论
中药尤其是复方中药,其功效是建立在多种复杂化学成分共同作用基础之上的,不能简单地以一种或几种成分来评价中药的有效性和专属性。中药的有效成分多数是次生代谢物,它们有很大的差异,同时具有一定的共性,很多成分出现在多种药材中,这对中药材的真伪鉴定和质量控制带来很大的难度。而中药指纹图谱是把中药材、中药制剂等作为研究对象,运用现代科学分析技术,找到能够表征其化学特征的色谱图或光谱图等数据;它是一种综合的、可量化的分析鉴定方法,是建立在中药复杂的化学成分系统研究的基础上,主要用于评价中药材、中药制剂、半成品质量的真实性、优良性和稳定性;它的显著特点是“整体性”和“模糊性”[13]。因此,对具有良好重复性的指纹图谱,用于中药材与中药制剂的质量控制,将对中药走向现代化和国际化具有重要意义。
采用全波长对参花胶囊样品进行测定,依据三维色谱图及对应波长下峰的信息量,结果254 nm时信息量较大,故将其作为HPLC指纹图谱的定性波长。测定4种化合物含量检测波长的选择,根据二极管阵列对绿原酸、羟基红花黄色素A、丹酚酸B、迷迭香酸扫描,结果最大吸收分别在326,403,286和329 nm,同时依据各峰的面积值与分离度(≥1.5),检测波长选择320 nm,各峰信息较为理想。
由于中药复方中成分众多,各成分溶解性不一样,等度洗脱难以充分洗脱,所以采用梯度洗脱的方法。选用Kromasil C18(250 mm×4. 6 mm,5 μm)色谱柱,采用梯度洗脱方法,比较了不同流动相系统乙腈-磷酸水溶液、甲醇-水、乙腈-醋酸水溶液、乙腈-水。考察了磷酸水溶液不同质量浓度(2.0,5.0,8.0和10.0 g·L-1)。结果表明,乙腈-5.0 g·L-1磷酸水溶液系统优于其他流动相系统,故选乙腈-5.0 g·L-1磷酸水溶液流动相系统作为本制剂指纹图谱流动相系统。
对制样方法进行了比较,采用甲醇与水超声提取,比较了甲醇的不同体积分数(20%,40%,50%,60%,70%,80%,90%和100%)、不同提取方法(超声、加热回流、浸渍等)、不同超声时间(5,10,15,20,30,40,50和60 min)。对不同溶剂比较,结果表明,甲醇优于水;对不同提取方法比较,结果表明,超声提取出峰多、峰面积大、时间短;对不同超声时间比较,结果表明,甲醇超声30 min以后,各个峰的面积不增加。综合上述各因素,选择70%甲醇、超声30 min,所得指纹峰较理想。
对不同柱温与流速进行比较,柱温超过或低于30 ℃,但大部分色谱峰分离度小或不能分开;流速高于或低于0.8 mL·min-1,大部分色谱峰分离度差。因此,选择便于控制的柱温30 ℃与流速0.8 mL·min-1。
综上所述,通过对10批次的参花胶囊样品进行相似度计算,结果数值介于0.966~0.998。同时,4种化合物定量结果,含量均一稳定,表明建立的参花胶囊的HPLC指纹图谱方法与含量测定方法,具有良好的分析评价能力,具有简便、稳定、可靠、准确等明显优势,因此,均可用于该制剂的质量控制。
参考文献:
[1] 程茜菲,刘银环,许苗苗,等. 丹参饮片HPLC指纹图谱研究[J]. 西北药学杂志,2013, 28(6): 556-558.
[2] 徐丽君,黄光英. 丹参的化学成分及其药理作用研究概述[J].中西医结合研究,2009,1(1):45-46.
[3] 刘海青,吕东,陈岳蓉. 全中药材市场商品丹参中4种丹参酮成分的HPLC测定[J]. 药物分析杂志,2005,25(2):229-230.
[4] 金玉青,洪远林,李建蕊,等. 川芎的化学成分及药理作用研究进展[J]. 中药与临床,2013,4(3) :44-48.
[5] 徐如英,童树洪. 红花的化学成分及药理作用研究进展[J]. 中国药业,2010,19(20):86-87.
[6] 田兰,吴桂荣,王岩. 红花黄色素研究进展[J]. 西北药学杂志,2007,22(4):218-220.
[7] 阮金兰,赵钟祥,曾庆忠,等. 赤芍化学成分和药理作用的研究进展[J]. 中国药理学通报,2003,19(9):965-970.
[8] 张清华,张玲. 菊花化学成分及药理作用的研究进展[J]. 食品与药品,2007,9(2):60-63.
[9] 吴士杰,李秋津, 肖学风,等. 山楂化学成分及药理作用的研究[J]. 药物评价研究,2010,33(4):316-319.
[10]刘育辰,陈有根,王丹. 甘草化学成分研究[J]. 药物分析杂志,2011,31(7):1251-1252.
[11]陶晡,刘晓清,屈振刚. 甘草化学成分研究进展[J]. 河北农业科学,2009,13 (3):77-79.
[12]中国药品食品监督管理局. 关于印发《中药注射剂指纹图谱研究的技术要求(暂行)》的通知)[J].中国药品标准,2000,1(4):3-4
[13]彭川丛,梁英娇. 中药指纹图谱研究进展[J]. 实用中医药杂志,2010,26(11):810-812.
猜你喜欢
中西医结合心脑血管病杂志(2022年12期)2022-07-07
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
中成药(2018年9期)2018-10-09
天然产物研究与开发(2018年7期)2018-08-21
中成药(2018年7期)2018-08-04
中成药(2018年2期)2018-05-09
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
