
类桃形钼酸铁微纳结构材料的水热合成、磁性和催化性能
2014-08-16 08:26邢春兰何广平南俊民
华南师范大学学报(自然科学版) 2014年3期
邢春兰,何广平,肖 信,南俊民
(华南师范大学化学与环境学院,广州510006)
钼酸铁(Fe2(MoO4)3)具有催化、光学和磁性等性质,在光致发光、微波传输、光学纤维、磁性材料、湿度传感器和催化等领域的应用前景受到广泛关注[1-6].前期研究中,通常采用高温固态反应合成Fe2(MoO4)3材料,但只能得到不均匀的微米粒状产品,这在一定程度上影响了材料的性能,限制了其在不同领域的应用.近期,研究者常用溶液化学法合成了具有特殊形貌和结构特征的Fe2(MoO4)3产品.例如,Zhang等[7]采用微波辅助液相合成法制备的分等级薄饼状Fe2(MoO4)3材料具有较强的光催化降解效果,对溴邻苯三酚红40 min的光降解率达到了65%左右;Kersen等[8]采用溶剂热法合成了无定型的Fe2(MoO4)3,并应用于选择性氧化H2S;Tian等[9]通过湿化学法合成了Fe2(MoO4)3类Fenton催化剂在中性条件下降解酸橙II(Acid Orange II),获得了较好的降解效果;此外,Fe2(MoO4)3也作为一种选择性较强的催化剂,应用于选择性氧化甲醇和丙烯等[10-12].因此,合理利用现代实验技术得到具有可控形貌和性能的Fe2(MoO4)3产品,也可以推动对该材料性能的认识,拓展材料的应用领域.
近期,课题组通过一步水热合成法,制备出了具有新颖分级结构、独特微/纳米结构的类桃形催化剂Fe2(MoO4)3材料,其制备工艺具有产品纯度高、分散性好、生产成本低、不需后续煅烧、结晶较好等特点.同时,该材料具有光催化和磁性双重性质,不仅对新型环境污染物(如盐酸四环素,TC)有优异的催化性能,还可以利用其磁性和沉降性能将该催化剂回收再利用.本文采用水热法合成了类桃形催化剂Fe2(MoO4)3材料,研究不同条件(反应时间、温度等)对材料形貌与结构、磁性及其降解TC的催化性能的影响.
1 实验方法
1.1 材料制备
制备Fe2(MoO4)3所用硝酸铁和钼酸铵均为分析纯试剂.首先,在超声辅助条件下,把2 mmol的Fe(NO3)3·9H2O 和 0.428 mmol的(NH4)6Mo7O24·4H2O分别溶解于15 mL去离子水中,得到Fe(NO3)3和(NH4)6Mo7O24溶液.然后,在磁力搅拌条件下,将(NH4)6Mo7O24溶液逐滴加入Fe(NO3)3溶液中,溶液由澄清逐渐变成乳黄色悬浮液,继续磁力搅拌30 min后,将黄色悬浮液转入以聚四氟乙烯为内衬的反应釜内(50 mL),放入恒温烘箱中进行水热反应.反应结束后,取出并打开反应釜,对产物进行离心分离,分别用去离子水和无水乙醇多次洗涤得到沉淀物,再在60℃下真空干燥即可得到产品.分别改变反应时间和温度,其他实验条件不变,即可得到不同反应条件的产品.
1.2 材料表征
采用D/Max-IIIA型全自动多晶X射线衍射仪(XRD;生产厂家日本Rigaku)测定Fe2(MoO4)3的晶体结构.测试条件为Cu K(λ=0.154 nm),工作电压30 kV,工作电流30 mA,扫描范围(2θ)为10~80°.使用JSM-6510-A型扫描电子显微镜(SEM;生产厂家日本JEOL)表征Fe2(MoO4)3的形貌.采用比表面积分析仪(BET)测定样品的孔径分布和比表面积,样品先在70℃真空干燥9 h,于77 K下进行氮气吸附-脱附测定.使用MPMS XL-7磁性能测试系统(生产厂家美国Quantum Design)检测材料的磁参数.
Fe2(MoO4)3的催化性能测试方法为,首先把TC(Aladdin公司)溶解到去离子水中配制成浓度为80 mg/L的TC溶液,取50 mL TC溶液放入250 mL烧杯中,加入50 mg Fe2(MoO4)3作为催化剂,在避光和一定温度条件下用磁力搅拌器中进行控温搅拌;然后,分别在反应进行 10、20、30、40、50、60 min时取出2.5 mL溶液,过滤后使用UV-1800型紫外-可见分光光度计(日本岛津)测定溶液在190~800 nm范围内的吸光度,根据最大吸收峰的变化检测溶液中TC的变化量,并绘制TC在不同温度条件下的降解曲线.TC的降解率(W)的计算公式为W=A0-At)/A0*100%,A0和At分别为TC溶液的初始及反应t时刻的吸光度.在避光条件下研究Fe2(MoO4)3对TC的催化降解性能,以排除光催化对测试结果的影响.此外,使用总有机碳分析仪(型号日本岛津)检测不同反应时间内溶液中总有机碳(TOC)的变化,以评估催化降解过程中有机物矿化的情况.
2 结果与讨论
2.1 制备的Fe2(MoO4)3形貌和结构
用SEM观察水热反应温度和时间对合成样品形貌的影响.图1A~D表明,随着反应温度的升高,产物形貌从无规则的片状转化为球状,当反应温度为180℃时,观察到产物粒径约为8μm均匀球状物.可见,反应温度对于产物最终形貌的构成起到了重要作用.由于不经过水热反应在自然沉降条件下得到的产品也为片状,因此180℃温度条件得到球状产物的过程中,必将经历一个物质形态的转换过程,即片状物质的溶解和再结晶形成球状产物.根据图2的XRD结果分析,水热前可以得到无定形的产物,而水热后可以得到具有较好晶型的产物,这可以从一个侧面支持溶解-再结晶机理.同时,从该桃形产物上有“桃脐”的出现则可作为发生溶解-再结晶过程的另一个证据.

图1 在100℃(A)、130℃(B)、160℃(C)和180℃(D)温度下水热反应30 min合成Fe2(MoO4)3样品的SEMFigure 1 SEM morphology of Fe2(MoO4)3 samples synthesized at100℃ (A),130℃ (B),160℃ (C)and 180℃(D)for 30 min

图2 180℃温度条件下水热反应不同时间制备Fe2(MoO4)3样品的XRD谱Figure 2 XRD patterns of Fe2(MoO4)3 samples synthesized at 180℃for different times
由于180℃温度条件下可以得到具有较好桃形的产物,故在该温度条件下考察了水热时间对产物形貌的影响(图3A~F).当反应时间超过0.5 h时,类桃形的球体产物外形则趋于稳定,同时,这些球体又是由二维结构的纳米片有序地层层堆积起来而构成球体.随着反应时间的增长,球体略微变大,晶体的外形结构得到进一步改善,“桃脐”消失,表明再结晶产物实际上依然存在溶解和再结晶过程,因形成比较规整的球状物可以使表面能量最小化,从而更加稳定.随着反应时间的延长(3 h,图3F),水热反应产物具有较大的外形和比较致密的组成.另外,从图3F中插图可以看出,3 h水热反应产物的内部具有较规则的内部纹理,该纹理则进一步支持了产物是在水热条件下通过再结晶逐步生成.

图3 180℃温度条件下水热反应0 min(A)、10 min(B)、30 min(C)、60 min(D)、120 min(E)和 180 min(F)得到样品的SEMFigure 3 SEM morphology of Fe2(MoO4)3 samples synthesizedat180℃ for 0 min(A),10 min(B),30 min(C),60 min(D),120 min(E)and 180 min(F)
将180℃温度条件下不同水热时间制备样品进行XRD分析(图2).未经过水热反应的产物为无定形状态,结晶性差.随着水热反应时间的增加,样品的结晶度逐渐增强,衍射峰宽度整体呈现由宽变窄趋势,表明样品的晶体尺寸增大和晶体的结晶更加完整.结晶良好的样品其衍射峰与单斜晶系Fe2(MoO4)3(JCPDS card no.35-0183)的衍射峰完全吻合,表明在合适的水热温度条件下可以得到单一相结构的产物.同时,随着水热时间的增加,样品的最强峰)和次强峰比值也在变化,说明在水热过程中Fe2(MoO4)3纳米晶体生长表现出各向异性生长,晶系不同晶面的生长速度不同.可见,从样品XRD图谱上反映出的信息,与从SEM图中得到的结果相吻合.此外,通过谢乐公式D=Kλ/βcosθ评估样品的晶粒尺寸,其中,λ是X射线辐射波长(λ =0.154 18 nm),K 是谢勒常数(K=0.89),θ是X射线衍射角,β是晶粒细化或微观应变引起的真实加宽.通过计算,反应30 min的样品,所得Fe2(MoO4)3的晶粒直径约为93 nm,表明产品是由纳米晶组成的微纳米结构.
采用N2吸附/解吸实验分析样品进行比表面积和孔径分布分析(图4),比表面积为145.149 3m2/g.Fe2(MoO4)3材料的N2吸附/解吸曲线属于IV型吸附等温线,且在较高压力时出现滞后环,这是由于样品存在孔状结构.在孔径分布曲线(图4B)中,在3 nm处左右出现了一个较宽的峰,这是因为“桃脐”形成时纳米条团聚挤压形成的.而其他空隙的直径从10 nm到100 nm不等,则是由组成桃形的纳米条相互堆叠时产生的空隙造成的.

图4 Fe2(MoO4)3材料的N2吸附/解吸(A)及BJH孔径分布(B)Figure 4 Nitrogen adsorption-desorption isotherms(A)and Barret-Joyner-Halenda(BJH)pore size distribution(B)of Fe2(MoO4)3
2.2 Fe2(MoO4)3的磁性能和催化性能
图5为180℃条件下水热反应1 h制备Fe2(MoO4)3样品的磁性数据.样品为磁性材料,其剩余磁感应强度(剩磁)、矫顽力与温度均无关.饱和磁场强度较小,可能与该材料较低的结晶度和小尺寸有关.当温度升高时,磁滞损耗变大.由于在5 K附近该磁性材料的矫顽力较小,且磁滞损耗较低,为软磁性材料.一般而言,矫顽力较小,磁滞损耗会比较大,但是当温度升高到300 K时,该材料的磁滞损耗却增大了,而矫顽力基本不变.因此,该磁性材料在低温下可应用于交流电器.

图5 Fe2(MoO4)3材料在5 K(A)和300 K(B)时的磁滞回线Figure 5 Hysteresis loops of Fe2(MoO4)3 at 5 K(A)and 300 K(B)
把Fe2(MoO4)3加入80 mg/L的TC溶液中,通过测量TC的降解率来评价所制备Fe2(MoO4)3的催化活性.催化实验结果表明,所制备样品对TC具有较好的催化降解活性,且催化活性与反应温度有关(图6).随着反应温度的提高,TC降解加快,70℃温度条件下反应50 min,TC的去除率约为90%,表明类桃形Fe2(MoO4)3微纳结构具有较高的催化活性,目标分子能够被降解.
为了研究Fe2(MoO4)3对有机污染物TC的真实去除(矿化)效果,对反应过程中溶液的总有机碳(TOC)进行了跟踪检测(图7).Fe2(MoO4)3微球对TC的催化降解作用明显,催化反应进行3 h后,TOC去除率为80%,意味着Fe2(MoO4)3材料能够有效矿化TC使其转化为无害的无机小分子.由于Fe2(MoO4)3具有较大的比表面积,对污染物吸附强,因此催化降解在材料的表面上进行.在材料表面上,Fe(Ⅲ)被还原,然后在空气中被迅速氧化,由于中间Fe(II)化合物被空气氧化的速度很快,因此在XRD图中没有检测到FeMoO4的存在.

图6 不同反应温度下Fe2(MoO4)3对TC的避光催化降解曲线Figure 6 Catalytic degradation of TC over Fe2(MoO4)3 at different temperatures in the dark condition

图7 Fe2(MoO4)3对TC的TOC降解率Figure 7 TOC removal ratio of TC over the Fe2(MoO4)3 microspheres
此外,Fe2(MoO4)3材料具有磁性并在液体中有较好的自沉降性,这可解决材料在应用中的回收分离问题.如图8所示,把Fe2(MoO4)3加入到TC溶液中,停止搅拌并使用磁铁进行吸附,Fe2(MoO4)3能够迅速全部沉降试管底部,这就解决了催化剂材料在反应结束后难于从固-液催化反应体系中回收的问题.
为了检验所合成Fe2(MoO4)3微球的稳定性和重复利用的性能,通过自然沉降分离出催化剂,再加入新鲜的TC溶液重新进行催化降解实验,如此反复3次,2 h后分别计算其降解效率(图9).Fe2(MoO4)3微球的降解效率下降了约7%,这是由循环和转移过程中催化剂的损失引起的.综合来看,Fe2(MoO4)3显示出良好的稳定性,这一点在实际应用中至关重要.

图8 TC溶液中的Fe2(MoO4)3材料沉降实验Figure8 Depositexperimentof the Fe2(MoO4)3 in TC aqueous solution,before Fe2(MoO4)3 adding(A),while adding(B)and aftermagnetic adsorption
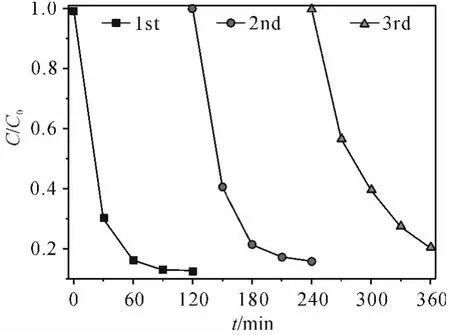
图9 Fe2(MoO4)3催化剂的回收性能Figure 9 Recycling properties of Fe2(MoO4)3 catalyst
3 结论
在低温、常压和没有添加模板剂或表面活性剂条件下,使用简单的水热法,采用不同实验条件成功合成了新型的分级Fe2(MoO4)3类桃状微球.对材料的结构和形貌形成机理进行了分析,认为反应温度对材料的形成起到了重要作用.通过时间序列实验,分析了Fe2(MoO4)3微球的形成过程,即自组装和溶解-重结晶机理.在避光条件下,以TC为模拟降解物,所合成Fe2(MoO4)3微球显示出优异的催化活性,并能够有效地矿化TC.材料具有磁性,可以很方便地实现固液分离回收,重复回收实验表明,所合成Fe2(MoO4)3材料具有很好的稳定性、容易回收且反复使用能够保持较高的活性,说明材料具有实际应用价值.
[1]Schmitt P,Brem N,Schunk S,et al.Polyol-mediated synthesis and properties of nanoscale molybdates/tungstates:Color,luminescence,catalysis[J].Advanced Function Materials,2011,21(16):3037-3046.
[2]Ryu JH,Yoon JW,Lim C S,et al.Microwave-assisted synthesis of barium molybdate by a citrate complexmethod and oriented aggregation[J].Materials Research Bulletin,2005,40(9):1468-1476.
[3]Sleight AW,Chamberland B L,Weiher JF.Magnetic,moessbauer,and structural studies on threemodifications of FeMoO4[J].Inorganic Chemistry,1968,7(6):1093-1098.
[4]KersenÜ,Menzel D.Study of themagnetic properties of themixed oxide Fe2O3-Fe2(MoO4)3in the temperature range 2-300 K with different compositions[J].Inorganica Chimica Acta,2011,368(1):96-100.
[5]SearsW M.The effectof oxygen stoichiometry on the humidity sensing characteristics of bismuth iron molybdate[J].Sensors and Actuators B,2000,67(2000):161-172.
[6]Andersson A,Henelind M,Augustsson O.A study of the ageing and deactivation phenomena occurring during operation of an iron molybdate catalyst in formaldehyde production[J].Catalysis Today,2006,112(1-4):40-44.
[7]Zhang L,Cao X F,Ma Y L,et al.Pancake-like Fe2(MoO4)3microstructures:Microwave-assisted hydrothermal synthesis,magnetic and photocatalytic properties[J].New Journal of Chemistry,2010,34(9):2027-2033.
[8]Kersen ü,Holappal L.Surface characterization and H2S-sensing potential of iron molybdate particles produced by supercritical solvothermal method and subsequent oxidation[J].Applied Physics A,2006,85(4):431-436.
[9]Tian SH,Tu Y T,Chen D S,etal.Degradation of Acid Orange IIatneutral pH using Fe2(MoO4)3as a heterogeneous Fenton-like catalyst[J].Chemical Engineering Journal,2011,169(1-3):31-37.
[10]Ivanov K,Dimitrov D,Boyanov B.Optimization of the methanol oxidation over iron-molybdate catalysts[J].Chemical Engineering Journal,2009,154(1-3):189-195.
[11]Ivanov K I,Dimitrov D Y.Deactivation of an industrial iron-molybdate catalyst formethanol oxidation[J].Catalysis Today,2010,154(3/4):250-255.
[12]Wang L,Ma T,ShengW,etal.Preparation,characterization,and properties of ferric molybdate nanotubes for propene epoxidation by air[J].Chinese Journal of Catalysis,2009,30(8):711-713.
猜你喜欢
军事文摘·科学少年(2017年4期)2017-06-20
材料科学与工程学报(2016年4期)2017-01-15
材料科学与工程学报(2016年2期)2017-01-15
光学精密工程(2016年2期)2016-11-07
科学大众·小诺贝尔(2016年6期)2016-08-17
中国光学(2015年1期)2015-06-06
应用化工(2014年8期)2014-08-08
郑州大学学报(工学版)(2014年6期)2014-03-01
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年8期)2014-02-28
