
G1对EA.hy926内皮细胞内质网应激的抑制作用
2014-08-15 06:50:44夏东晖曹幸毅王静宇袁明吴士文
中国实验动物学报 2014年2期
夏东晖,曹幸毅,王静宇,袁明,吴士文
(1.新乡医学院,新乡市 453003;
2.中国人民武装警察部队总医院,北京市 100039;
3.中国航天员科研训练中心,航天医学基础与应用国家重点实验室,北京市 100094)
内皮细胞损伤是动脉粥样硬化(atherosclerosis,AS)发生的始动因素之一,持续的高血糖可诱导血管内皮细胞损伤甚至凋亡[1]。内质网应激(endoplasmic reticulum stress,ERS)是细胞应对有害刺激时的一种保护性调节机制,但持续或程度较强的ERS可引起血管内皮细胞损伤及功能障碍,从而促进动脉粥样硬化的发生[2-3]。先前的研究表明,雌激素可通过对抗高糖诱导的ERS调节内皮细胞功能[4]。GPR30作为新近发现的雌激素受体,激动GPR30可抵抗高糖导致的内皮细胞凋亡[5],但这种保护作用是否通过抑制ERS相关通路尚未被阐明。本实验应用高浓度葡萄糖干预EA.hy926内皮细胞,观察ERS与高糖诱导内皮细胞凋亡的关系,研究GPR30的特异性受体激动剂G1对高糖诱导内皮细胞ERS及凋亡的影响,探讨其保护效应。
1 材料与方法
1.1 试剂与仪器
主要试剂:GPR30受体激动剂G1(Sigma 公司, 美国),Annex V细胞凋亡检测试剂盒(BD Pharmingen公司, 美国), 抗Bip、IRE1、PERK、Bax、Bcl-2抗体(Cell Signaling Technology公司, 美国),Trizol(Invitrogen公司, 美国)、PCR引物(北京奥科生物技术有限公司,中国),反转录及实时定量PCR试剂盒(TaKaRa 公司, 日本)。主要仪器:二氧化碳细胞培养箱(NUAIRE公司, 美国),倒置显微镜(Nikon TS100,日本),实时定量PCR仪(Eppendorf 公司, 美国),电泳系统(Bio-rad, 美国),流式细胞仪(BD FACS Aria II, 美国)。
1.2 方法
1.2.1 细胞分组及处理方法
EA.hy926内皮细胞培养于含10%胎牛血清、100U/mL青霉素、100U/mL链霉素的DMEM/F-12培养液中,在5%CO2、37℃细胞培养箱中培养。细胞达80%融合状态时,将细胞分为3组:正常对照组(Con,17.51 mmol/L 葡萄糖)、高糖组(HG,33.3 mmol/L)、高糖+G1组(HG+G1,HG+1μmol/L G1),3组细胞同时处理24 h。
1.2.2 流式细胞术检测细胞凋亡[6]
根据实验分组处理后,使用0.25%胰酶消化EA.hy926内皮细胞,PBS清洗1次,1500 g离心5 min,弃上清,收集细胞(1×106细胞/组)。PBS洗3次,以结合缓冲液重悬细胞沉淀,使细胞浓度为1×106/mL,100 μL细胞悬液,加入annexinV-PE及7-AAD各5 μL,室温避光15 min,加入400 μL结合缓冲液,进行流式细胞术定量检测。以不加annexinV-PE及7-AAD的一管作为阴性对照。凋亡及死亡的细胞膜磷脂酰丝氨酸外翻以致annexin V染色阳性;而活细胞annexin V染色阴性;死亡细胞则为7-AAD染色阳性。即第一象限7-AAD +/annexin V+为死亡细胞,第二象限7-AAD +/annexin V-为机械损伤的细胞,第三象限7-AAD-/annexin V-为活细胞,第四象限7-AAD -/annexin V+为凋亡细胞。
1.2.3 RNA提取与实时定量PCR
按实验分组处理EA.hy926内皮细胞并用Trizol法提取各组总RNA,取RNA产物2 μg反转录合成cDNA。PCR反应条件:95℃预变性30 s,95℃变性5 s,60℃退火30 s,循环40次。使用Eppendorff mastercycle荧光定量PCR仪进行扩增及定量分析(表1)。

表1 PCR引物序列
1.2.4 蛋白印迹法[6]检测ERS相关分子蛋白表达
根据实验分组处理EA.hy926内皮细胞,24 h后收集细胞(1×106细胞/组),使用蛋白裂解液及超声法提取细胞总蛋白,BCA法测蛋白浓度,取30 μg蛋白加入样品缓冲液,100℃水煮沸5 min。配制SDS-PAGE凝胶后加入样品后进行电泳,电泳结束后通过半干电转仪将样品转移到PVDF膜上,室温下摇床用含5%脱脂奶粉的TBS-T封闭1 h,以Bip、IRE1、PERK、Bax、Bcl-2、与抗体稀释液(1∶1000)分别4℃孵育过夜。TBS-T清洗PVDF膜3次,每次10 min,然后与HRP抗兔抗体(1∶1000)室温孵育1 h后,TBS-T清洗PVDF膜3次,每次10 min,ECL显色系统X线感光显影。用扫描仪对图片进行扫描,以GAPDH蛋白作为内参照,进行半定量分析。
1.2.5 统计分析

2 结果
2.1 高糖对EA.hy926内皮细胞凋亡的影响及G1的作用
HG组与Con组比较,细胞凋亡率明显升高[(11.2±0.2)% vs(5.4±0.18)%,P<0.01],差异有显著性;HG+G1组与HG组比较,细胞凋亡率明显降低[(7.8±0.15)% vs (11.2±0.2)%,P<0.05],差异有显著性。见图1。

注:**为HG组与Con组相比差异有显著性,P<0.01;*为HG+G1组与HG组相比差异有显著性, P<0.05。
2.2 高糖对EA.hy926细胞凋亡相关分子和ERS相关蛋白表达的影响及G1的作用
HG组与Con组比较,Bip、IRE1、PERK及凋亡分子Bax表达上调(P<0.05,P<0.01或P<0.001),Bcl-2的表达下调(P<0.01),差异有显著性;HG+G1组与HG组比较, Bip、IRE1、PERK及凋亡分子Bax表达下调(P<0.05或P<0.01),Bcl-2的表达上调(P<0.05)差异均有显著性。见图2。
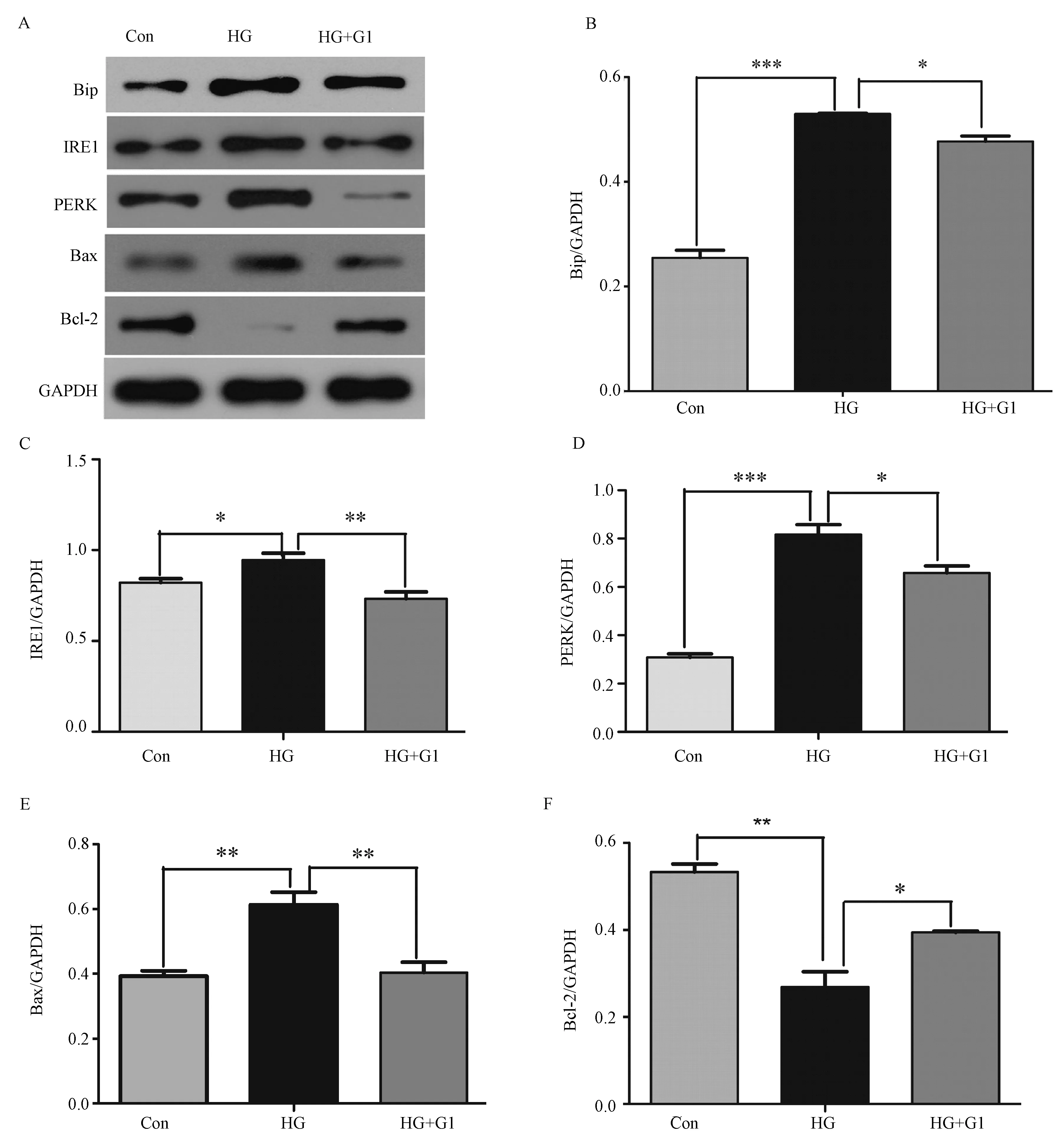
注:HG组与Con组相比,Bip表达上调(P<0.001);IRE1表达上调(P<0.05);PERK表达上调(P<0.001);Bax表达上调(P<0.01); Bcl-2表达下调(P<0.01)。结果差异均有显著性。HG+G1组与HG组相比,Bip表达下调(P<0.05);IRE1表达下调差异有显著性(P<0.01);PERK表达下调(P<0.05); Bax表达下调(P<0.01);Bcl-2表达上调(P<0.05)结果差异均有显著性。
2.3 高糖对内皮细胞Bip mRNA和CHOP mRNA表达的影响及G1的作用
HG组较Con组,Bip mRNA、CHOP mRNA 表达上调(P<0.001及P<0.01),差异有显著性;HG+G1组与HG组比较,Bip mRNA、CHOP mRNA 表达下调(P<0.001及P<0.01),差异有显著性。见图3。
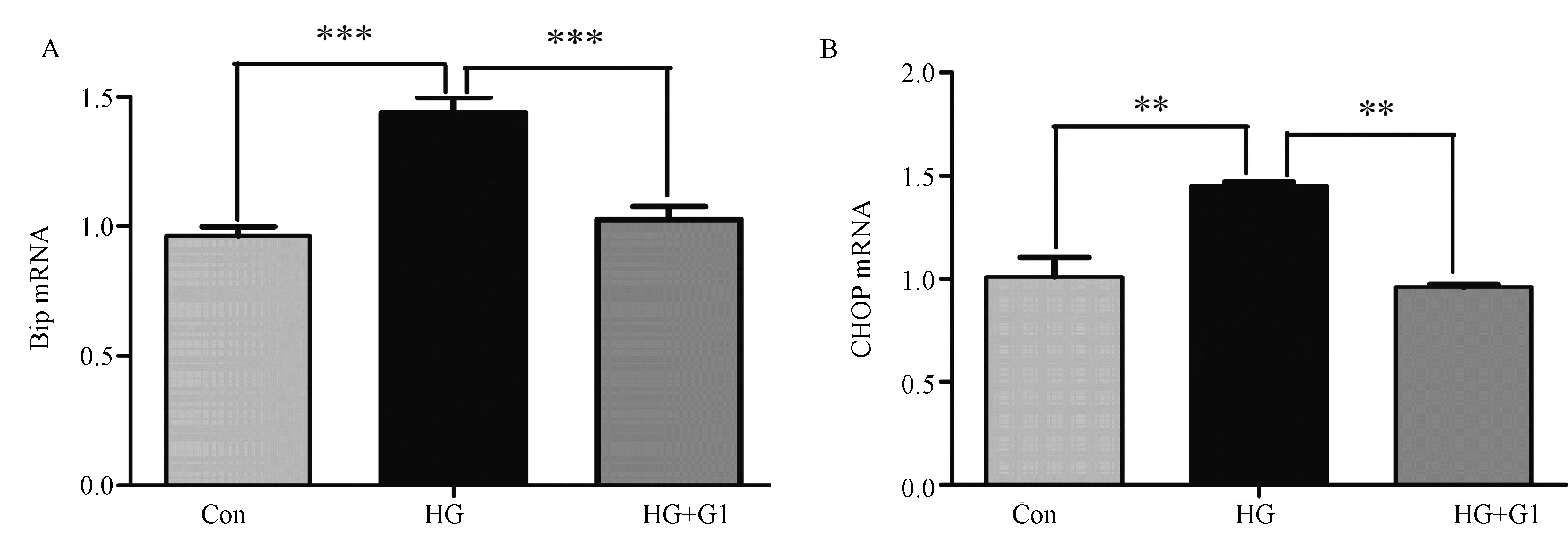
注:***为HG组与Con组相比,HG+G1组与HG组相比,BIP mRNA表达变化差异有显著性,P<0.001; **为HG组与Con组相比,HG+G1组与HG组相比,CHOP mRNA表达变化差异有显著性, P<0.01。
3 讨论
血管内皮细胞损伤是动脉粥样硬化发生的早期事件,致动脉粥样硬化的危险因素均可导致血管内皮细胞损伤甚至细胞凋亡[7]。已有研究报道,高糖作为动脉粥样硬化的危险因素,可通过诱导内皮细胞损伤促进动脉粥样硬化的发生发展[8],本研究通过流式细胞术证实持续的高糖可诱导EA.hy926内皮细胞凋亡。
细胞凋亡主要有三条通路:死亡受体活化通路、线粒体损伤通路和内质网应激启动的凋亡通路,前两者是经典凋亡通路,而内质网应激通路是近年来才发现的一种新的凋亡途径[9]。内质网是真核细胞中蛋白质折叠加工的主要场所。由于各种原因引起的内质网中出现错误折叠与未折叠蛋白在腔内聚集以及Ca2+平衡紊乱的状态称为内质网应激。Bruno等[10]的研究表明,持续的高血糖可上调内质网应激相关蛋白Bip、PERK、IRE1,Bip是内质网中一种特征性的伴侣蛋白,其诱导表达对内质网应激起着关键作用,被认为是内质网应激信号系统上游的开关分子,是内质网应激的一种标志蛋白[11]。PERK、IRE1在正常情况下都以无活性的状态与分子伴侣Bip结合,内质网应激时未折叠蛋白的聚集使Bip与这两种蛋白分离[12],本实验结果表明,持续的高糖可上调EA.hy926内皮细胞ERS的标志蛋白Bip,上游的感受器蛋白PERK、IRE1。PERK、IRE1都能诱导转录因子CHOP的表达,在生理状况下,CHOP的表达量很少,主要存在于细胞质中,而当细胞发生内质网应激时,CHOP的含量大量增加并聚集在细胞核内[13]。本实验结果表明,HG组较Con组,ERS特异的转录因子CHOP mRNA表达增加,具有显著差异,过量表达的CHOP通过激活GADD34、ERO1和死亡受体DR5等凋亡反应蛋白促进细胞凋亡,也能调节其他基因的转录,比如Bcl-2家族蛋白,Bcl-2家族在细胞凋亡领域中倍受关注,细胞对凋亡刺激的敏感性很大程度上取决于Bcl-2家族促凋亡与抗凋亡蛋白的平衡。CHOP与cAMP反应元件结合蛋白(CREP)形成二聚体能抑制Bcl-2蛋白的表达,进而促进线粒体对促凋亡因素的敏感性[14]。本实验HG组与Con组比较,凋亡蛋白Bax表达增加,抗凋亡蛋白Bcl-2表达降低。正常血管内皮细胞中Bax蛋白表达较低,当细胞受到损伤后Bax蛋白表达增加,Bcl-2蛋白表达降低,对细胞凋亡的发生和调节发挥重要作用[15],因此,持续的高糖可促进EA.hy926内皮细胞凋亡,从而促进动脉粥样硬化的发生。
雌激素是类固醇激素家族中重要的激素,参与调节许多生理与病理效应,具有重要的心血管保护作用,主要表现在保护内皮、抑制血管平滑肌细胞增殖迁移、减少动脉粥样硬化斑块形成和促进斑块消退等方面[16]。Haas等[4]的研究证实,雌激素可通过对抗高糖诱导的ERS调节内皮细胞功能,GPR30作为新近发现的雌激素受体,Li等[5]的研究表明,激动GPR30可抵抗高糖导致的内皮细胞凋亡,但此作用是否通过ERS相关通路尚未见报道。本实验以高糖诱导EA.hy926内皮细胞ERS为模型,证实了GPR30特异性受体激动剂G1可下调高糖诱导的ERS相关蛋白Bip、PERK、IRE1以及凋亡分子Bax,CHOP mRNA,也可上调抗凋亡蛋白Bcl-2的表达。因此,GPR30特异性受体激动剂G1能对抗高糖诱导的ERS及细胞凋亡,从而拮抗动脉粥样硬化的发生。
综上所述,持续高糖诱导的内皮细胞凋亡作用,部分是因为激活了ERS相关凋亡信号通路,GPR30特异性受体激动剂G1可抑制EA.hy926内皮细胞内质网应激从而发挥抗凋亡作用。
[1] Su Y, Mao N, Li M et al.Sarpogrelate inhibits the expression of ICAM-1 and monocyte-endothelial adhesion induced by high glucose in human endothelial cells[J].Mol Cell Biochem, 2013,373(1) :195-199.
[2] Sanson M, Auge N, Vindis C, et al.Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells:prevention by oxygen-regulated protein 150 expression[J].Circ Res,2009,104(3):328-336.
[3] Kawanami D, Matoba K, Okada R, et al.Fasudil inhibits ER stress-induced VCAM-1 expression by modulating unfolded protein response in endothelial cells [J].Biochem Biophys Res Commun, 2013,435(2):171-175.
[4] Haas MJ,Raheja P,Jaimungal S.Estrogen-dependent inhibition of dextrose-induced endoplasmic reticulum stress and superoxide generation in endothelial cells[J].Free Radic Biol Med.2012,52(11):2161-2167.
[5] Li ZL, Liu JC,Liu SB,et al.Improvement of vascular function by acute and chronic treatment with the GPR30 agonist G-1 in experimental diabetes mellitus[J].Plos One, 2012, 7(6):e38787.
[6] 贾方,孙建辉,吴春芳,等.阿托伐他汀对同型半胱氨酸诱导内皮细胞内质网应激的抑制作用[J].江苏大学学报(医学版)2012,22(6):483-490.
[7] Gimbrone MA Jr, Garcia-Cardena G.Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis [J].Cardiovasc Pathol.2013,22(1):9-15.
[8] Schisano B, Harte AL, Lois K, et al.GLP-1 analogue, Liraglutide protects human umbilical vein endothelial cells against high glucose induced endoplasmic reticulum stress[J].Regul Pept, 2012,174(3):46-52.
[9] Verfaillie T, Garg AD, Agostinis P et al.Targeting ER stress induced apoptosis and inflammation in cancer [J].Cancer Lett.2013, 332(2):249-264.
[10] SU Y C,WU J L,HONG J R.Beta nodavirus up-regulates chaperone GRP78 via ER stress:role of GRP78 in viral replication and host mitochondria -mediated cell death[J].Apoptosis,2011,16(3):272-287.
[11] Sasaki S, Samejima S, Uruga T, et al.Synthetic studies of the spirocyclic cyclohexene part of versipelostatin, a novel GRP78/Bip molecular chaperone down regulator[J].Antibiot (Tokyo).2013, 66(3) :147-154.
[12] Huber AL, Lebeau J, Guillaumot P, et al.p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose[J].Mol Cell.2013,49(6) :1049-1059.
[13] Wu XD, Zhang ZY, Sun S, et al.Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress [J].Apoptosis, 2013,18 (1):85-98.
[14] Dalton LE,Clarke HJ,Knight J,et al.The endoplasmic reticulum stress marker CHOP predicts survival in malignant mesothelioma[J].Br J Cancer, 2013,108(6):1340-1347.
[15] 王彪, 庄福连, 张鹏飞, 等.细胞凋亡及Bcl-2、Bax在血管瘤和血管畸形中的表达 [J].中华整形外科杂志 2003, 19(5):347-349.
[16] Munir JA,Wu H,Bauer K,et al.The perimenopausal atherosclerosis transition: relationships between calcified and noncalcified coronary, aortic, and carotid atherosclerosis and risk factors and hormone levels [J].Menopause.2012,19(1):10-15.
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18 03:53:24
现代临床医学(2021年1期)2021-01-26 00:55:52
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:18:21
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
长江蔬菜(2015年3期)2015-03-11 15:10:29
中国当代医药(2015年33期)2015-03-01 02:09:08
