
Ga掺杂改性ZnO(001)面对CO气敏机理的第一性原理计算*
2014-08-13 11:33:30于灵敏韦建松范新会
西安工业大学学报 2014年6期
于灵敏,韦建松,张 荣,雷 曼,范新会,严 文
(西安工业大学 材料与化工学院,西安 710021)
ZnO对CO的气敏机理模型最为广泛是表面氧吸附模型.该模型主要描述了这样的一个过程:用ZnO作为传感材料对CO的识别首先是由表面发生氧吸附开始的.吸附的氧首先是以物理吸附的形式存在于ZnO表面,当其获得一定的能量后,进入化学吸附形式.在这个过程中空气中的氧夺取表面电子变成化学吸附氧,吸附氧变为氧离子,电子将从表面转移到氧中,从而导致表面电子浓度降低,电阻升高.这种吸附的氧离子作为电子受主态存在于带隙中;当环境气氛中存在还原性CO气体时,预吸附的氧就与CO气体在气敏材料表面发生反应,电子将释放回导带,使得ZnO材料电阻降低,从而达到测试气体的作用[1].尽管这个模型对指导n型半导体氧化物的氧吸附很有作用,但它也有一定的局限性.它不能为吸附氧种类的位置和几何形状,电子转移数目等提供参考[2].
目前,第一性原理计算已广泛应用于材料性质和材料设计的计算之中,是目前较为准确的电子结构计算的理论方法[3-5].Hu Ming[6-7]等用第一性原理计算了Ti掺杂 WO3(002)面对NO2和NH3的气敏机理.Benjawan Kaewruksa[8]等用第一性原理计算了ZnO吸附小分子气体的气敏性能.J.S.Michelle[9]等综述了一维ZnO纳米材料的气敏性能的第一性原理计算.Javad Beheshtian[10]等用第一性原理计算了ZnO纳米团簇吸附Cl2的气敏机理.Jakub Soltys[11]等用第一性原理计算了 ZnO(0001)和(000)极性面对 Zn,O2和 O 的吸附特性.M.Breedon[12]等用第一性原理计算了ZnO()吸附NO和NO2的气敏机理.但是,迄今为止,还未见到Ga掺杂改性ZnO(001)面对CO气敏机理的第一性原理计算的相关文献.
文中计算过程中采用Materials Studio计算模拟软件中的Castep第一性原理计算软件包,研究Ga(6.25mol%)掺杂的ZnO对CO气体吸附的影响,建立相应的模型,从理论上研究Ga掺杂改性ZnO对CO的气敏机理.
1 理论模型和计算方法
文中主要的计算工作由Materials Studio材料计算模拟软件中的Castep软件包完成.计算过程中采用周期性边界条件,利用广义梯度近似(Generalized Gradient Approximation,GGA)的 Perdew-Burke-Ernzerhof(PBE)方法来处理电子间的交换关联能.为了尽量减少平面波基矢个数,采用了超软赝势来描述离子实与价电子之间的相互作用势,选取 O—2s22p4、Zn—3d104s2、Ga—3d104s24p1组态电子作为价电子,其余轨道电子被视为芯电子进行计算.
在常温常压下稳定的ZnO是六方钎锌矿结构,属于P63mc空间群,对称性C6V-4,晶格常数a=b=0.325nm,c =0.5207nm,α=β=90°,γ=120°,c/a为1.602,较理想的六角柱密堆积结构的1.633稍小.晶胞由氧的六角密堆积和锌的六角密堆积沿c轴平移0.382反向套构而成.每个Zn原子与最近邻的四个O原子构成一个四面体结构.同样,每个O原子和最近邻的四个Zn原子也构成一个四面体结构.四面体并非严格对称,在c轴方向上,Zn原子与O原子之间的距离为0.196nm,而在其它三个方向上则为0.198nm,Zn-O键是典型的sp3杂化.其2×2×2超晶胞结构分别有16个O原子和16个Zn原子,文中所有计算采用此模型.
2 结果与分析
2.1 CO吸附在ZnO表面的晶体模型
ZnO体结构模型建立之后,由于所做研究是表面电子结构,所以需要先切面.计算过程中发现ZnO四个表面吸附能的排列为:(110)< (100)<(101)< (001).因此,我们预测,(110)面是这四个面中最稳定的;相反,(001)则是最不稳定的.因此,我们选择(001)面作为CO的吸附表面.
采用广义梯度近似,首先对构建的(001)结构进行了优化.其动能截止能量为340eV,K点取值为4×4×2.在此条件下进行赝势和电荷密度的自洽迭代循环.迭代过程中的收敛精度为1×10-5eV,作用在每个原子上的力不大于0.03eV/nm,内应力不大于0.1GPa.
为避免所选取的表面薄层结构优化时两个面发生弛豫而影响对表面吸附的研究[13],下面的原子层被固定,最上层的Zn和O原子允许发生弛豫.对纤锌矿晶体2×2×1超晶胞的结构进行优化,即计算得到电子能量的最小化和几何结构稳定化.图1为经过几何优化后CO吸附在ZnO(001)面的晶体模型.
经过优化后表层原子的位移、键长等发生了变化.这表明该表面优化后的结构发生了一定的变化.优化后的CO自由分子C-O键长为0.1153 nm,与实验测得的0.1129nm非常接近.C原子与ZnO表面发生相互作用,导致C-O键强度减弱,即CO已被活化.从键长角度分析,C-O键改变越多,则该基体对CO分子的活化作用就越强.CO分子键长增长,这说明CO分子内碳氧原子库仑排斥在增强,它们的结构也越不稳定,与吸附能计算结果一致.由于CO分子之间的排斥作用和ZnO表层原子的吸引,CO分子并不平行轴,而发生了偏移.
用公式来计算掺杂前后的吸附能Eads,即
问:那么您刚刚提到“理论商店”,我想很多研究者在研究的过程中都会不可避免地和理论产生交集,尤其是年轻研究者可能会苦于还没有找到一个合适的解释理论,或者在了解理论的基础上苦于找寻不到自己明确的研究问题,那么您是如何定义研究中生成的理论呢?以及一个理论在研究中的作用呢?

式中:Esubstrate-adsorbate为吸附 -基体的总能 量;Esubstrate和Eadsorbate分别是基体ZnO和吸附CO的能量.计算结果显示Eads(未掺杂)=8581eV,Eads(掺杂)=8751eV.正值对应的是放热过程,表示一个稳定的吸附.利用理想模型计算的吸附能并不能与实际实验中的吸附能量相比,但作为定性对比分析同一系统的吸附性能是可行的.

图1 CO吸附在ZnO(001)的晶体模型Fig.1 The crystal model of ZnO(001)attached by CO
2.2 本征ZnO气敏机理的第一性原理计算
2.2.1 本征ZnO的能带结构与电子结构
图2是本征ZnO的能带结构,G点是对称点,其宽带间隙为0.726eV,它的费米能级在导带和价带之间,可以看出本征ZnO是一种直接禁带半导体,导带底和价带顶位于Brillouin区的G点处.

图2 本征ZnO的能带结构图Fig.2 The band structure of intrinsic ZnO
图3是ZnO的态密度和分态密度图.纯净的ZnO是绝缘体,费米能级在禁带的中部附近,可以看出本征ZnO价带可以分为两个部分,即位于-6.6~-2.6eV的下价带和位于-2.6~0eV的上价带.上价带主要是由O 2p态贡献,下价带主要是由Zn 3d态所构成,导带主要是由Zn 4s和O 2p态所贡献,而且电子具有明显的从Zn 4s态到O 2p态的跃迁过程,引起O位置处的局域态密度的引力中心向低能级方向移动,表明ZnO是一个离子性较强而共价性较弱的混合键金属氧化物半导体.计算得到的本征ZnO的能带间隙为0.726eV,远远小于实验值3.37eV.这个差异是由计算过程中所采用的GGA近似所造成的.

图3 本征ZnO的态密度及分态密度图Fig.3 State density and part state density of intrinsic ZnO
2.2.2 本征ZnO(001)吸附CO的气敏机理的第一性原理计算
在吸附模型的气敏机理中认为,由于气体吸附作用,吸附气体会在氧化物表面捕获或释放电子而使表面电子性能发生改变,影响其电导变化.为了进一步考察ZnO(001)面吸附CO后的电子性能的变化,对吸附后的电子结构和能带结构进行了计算.图4~5分别为ZnO吸附CO后的能带结构和态密度.

图4 ZnO(001)吸附CO后的能带结构Fig.4 The band structure of ZnO(001)attached by CO
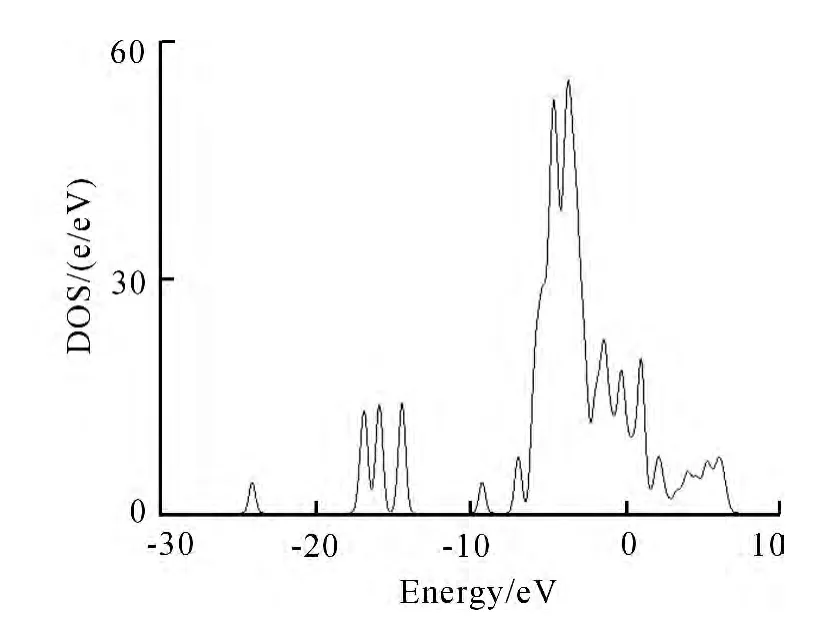
图5 ZnO(001)吸附CO后的态密度Fig.5 The state density of ZnO(001)attached by CO
吸附对整体表面的总电子态密度分布没有产生明显改变,价带与导带部分态密度电子峰强及宽度等未发生改变,只是在-20~-15eV出现了几个小峰,这是由于C原子和ZnO表面的Zn原子发生反应.总的来说,价带部分色散加剧而且展宽.导带处的态密度强度增加.这主要是由于吸附CO后,ZnO表面获取了CO的电子,使得空穴电子浓度增大,因此费米能向高能位移动,导电性增加.这也说明了CO与ZnO(001)面发生了明显作用.
2.3 Ga掺杂ZnO气敏性能提高的第一性原理计算
图6是一个Ga原子替换超晶胞中的锌原子的能带结构(即掺杂浓度为6.25mol%的ZnO),其宽带间隙为0.5302eV,可以看出费米能级向导带方向移动,能带向低能方向移动,能带发生简并.
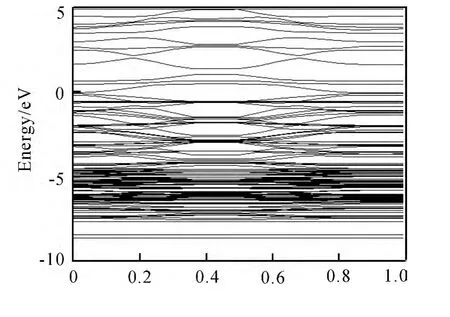
图6 Ga掺杂ZnO未吸附CO的能带结构图Fig.6 The band structure of Ga-doped ZnO without attached CO
图7是Ga掺杂ZnO未吸附CO的总态密度和分态密度图.
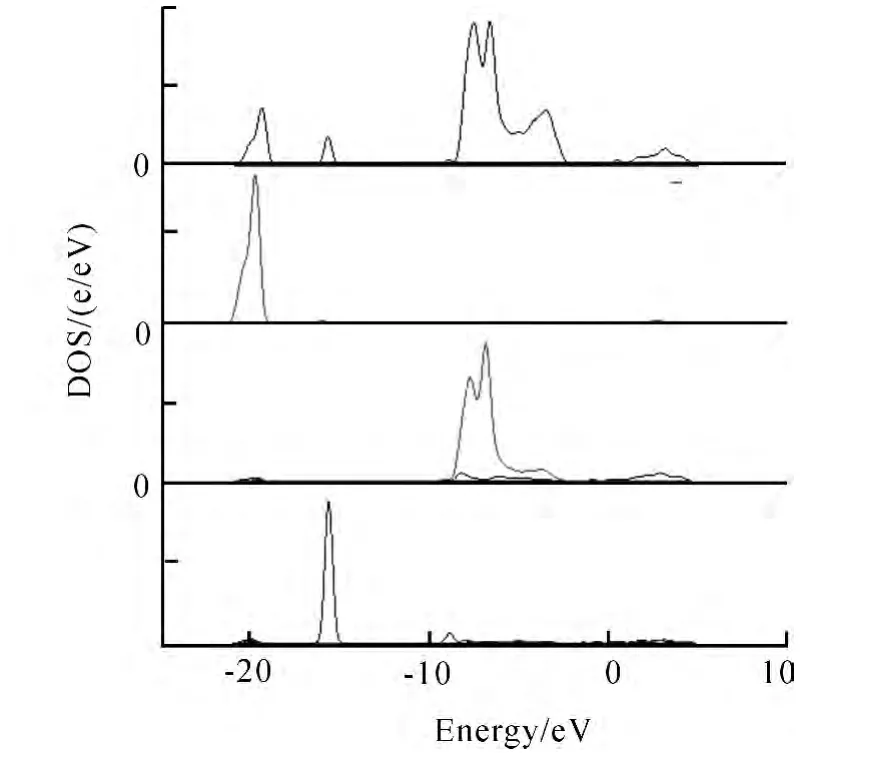
图7 Ga掺杂ZnO未吸附CO的总态密度图Fig.7 Total state density of Ga doped ZnO without attached CO
从图7中可以看出掺杂Ga后总态密度稍微发生了变化,在Zn 3d态占主导下价带引入了宽度约为0.1eV的由掺杂原子Ga 4s贡献的电子态.与没有掺杂的态密度图5比较,态密度向低能方向移动,这主要是由于高浓度掺杂产生的自由载流子,从以下两个方面改变ZnO材料的带隙:①是高浓度载流子使费米能级移入导带而产生所谓的Burstein-Moss[14]移动,使光学吸收边向低能方向移动而使带隙变宽;②电荷之间相互作用产生多体效应或杂质及缺陷带之间的重叠使带隙变窄.但前者的作用小于后者.在-17eV处还出现了宽度约为1.5eV由d态贡献的能带,其它价带部分与没有掺杂的情况大体相同.但与未掺杂情况相比,价带部分色散加剧,这主要是在高浓度掺杂情况下,杂质原子间互相很靠近,被掺杂离子束缚的电子波函数显著重叠,造成掺杂原子s轨道与O 2p轨道电子共有化运动加剧,展宽了价带部分.Ga掺杂对ZnO导带部分的影响,由图7的分波态密度的对比可以看出:Ga对导带态密度的贡献明显大于Zn2+对导带态密度的贡献;掺杂后导带表现出高度的弥散性,几乎没有局域化的特征.对于进入导带的费米能级处的态密度主要来源于掺杂原子的贡献,而Zn原子对其的贡献很小,在导带底部存在大量的自由电子,可见Ga掺杂不仅对价带有影响还对导带有贡献,提高了ZnO的导电性.
2.3.2 Ga掺杂ZnO吸附CO的能带结构与电子结构
在计算电子结构中我们发现随着掺杂浓度增大,体系的总能量在升高说明Ga3+取代Zn2+后晶体的稳定性在降低.掺杂Ga后的ZnO(001)面原子移动方向与未掺杂的ZnO(001)面一致,而键长有所变化.计算掺杂前后的吸附能,发现掺杂后的(001)面吸附能相对未掺杂前增大,由此可初步确定,掺杂Ga后的(001)面可产生更多的吸附气体,有利于测试气体的气敏反应.
气敏测试表明,掺杂微量的Ga提高了ZnO对测试气体的气敏性,而对其掺杂前后的材料结构进行表征后,发现掺杂Ga并未改变ZnO的晶体结构,粒子尺寸及形貌等并未发现有明显改变.为了揭示出Ga如何对ZnO的气敏性能产生影响,在此根据图1中的Ga掺杂ZnO的原子模型,然后计算出ZnO(001)面吸附CO后的能带结构和态密度,图8~9分别是Ga掺杂ZnO吸附CO的能带结构和态密度图.
掺杂Ga后ZnO的(001)面吸附CO分子后的总电子态密度分布与掺杂前类似,带隙中未出现其他的电子态,说明表面仍然保持半导体的性质.尽管图形结构基本一致,但掺杂后的ZnO表面对CO吸附前后的电子态密度发生了显著的变化,即我们主要考虑的费米能级的位置发生了很大变化.随着Ga原子的掺杂,价带和导带之间的间隙变小了,费米能级发生了明显的改变进入了导带.价带部分色散加剧而且展宽,导带处的态密度强度增加.这是由于吸附CO具有很强的给电子能力,当吸附于ZnO(001)面后,电荷会从CO中转移到ZnO(001)面的原子中,表面空位电子浓度增加.因此在导带底部由于CO给出的电子而形成的表面态能级增强,使得费米能级向高能位移动.另外,Ga原子是施主掺杂,促使费米能级进入导带处,从而载流子浓度增加.

图8 Ga掺杂ZnO吸附CO后的能带结构Fig.8 The band structure of Ga doped ZnO(001)attached by CO

图9 Ga掺杂ZnO吸附CO后的态密度Fig.9 The state density of Ga doped ZnO(001)attached by CO
总之,随着Ga浓度的增加,ZnO的能带隙逐渐减小,费米能级向导带移动,更多的载流子受到激发从价带跃迁到导带上.Ga的掺杂在半导体表面产生更多的电子,使其电阻率下降,吸附更多的气体分子,显示更优异的气敏特性.
3 结论
文中基于第一性原理计算了纯ZnO和Ga掺杂的ZnO的(001)面吸附CO分子后的电子结构和能带结构,对Ga掺杂改性ZnO(001)面吸附CO的气敏机理进行了理论解释.研究结果表明纯ZnO(001)吸附CO后,由于C原子和ZnO表面的Zn原子发生反应,在-20~-15eV出现了几个小峰,但是吸附对整体表面的总电子态密度分布没有产生明显改变,价带与导带部分态密度电子峰强及宽度等未发生改变.而掺杂Ga后ZnO的(001)面的总电子态密度分布与掺杂前类似,带隙中未出现其他的电子态,但Ga掺杂后的ZnO表面吸附CO后,电荷从CO转移到Ga掺杂的ZnO(001)面原子中,导致表面空位电子浓度增加,价带和导带之间的间隙变小,费米能级进入导带.因此Ga掺杂改性的ZnO(001)将在ZnO表面产生更多的电子,使其电阻率下降,吸附更多的CO气体分子,显示更优异的气敏特性.
[1]YU L M,FAN X H,CAO L,et al.Gas Sensing Enhancement of Aluminum-doped ZnO Nanovase Structure with Many Gas Facile Diffusivity Paths[J].Applied Surface Science,2013,265:108.
[2]曾文,刘天模.SnO2/TiO2体系气敏性能及其机理研究[D].重庆:重庆大学,2011.ZENG Wen,LIU Tian-mo.Study on Gas Sensing Properties and Mechanism of SnO2/TiO2Systems[D].Chongqing:Chongqing University,2011.(in Chinese)
[3]刘建军.掺Ga对ZnO电子态密度和光学性质的影响[J].物理学报,2010,59(9):6466.LIU Jian-jun.The Effect on Electronic Density States and Optical Properties of ZnO by Doping of Ga[J].Acta Physica Sinica,2010,59(9):6466.(in Chinese)
[4]张富春,邓周虎,阎军锋,等.ZnO电子结构与光学性质的 第 一 性 原 理 计 算 [J].光 学 学 报,2006,26(7):1203.ZHANG Fu-chun,DENG Zhou-hu,YAN Jun-feng,et al.First-Principles Calculation of Electronic Structure and Optical Properties of ZnO[J].Acta Optica Sinica,2006,26(7):1203.(in Chinese)
[5]PRADES J D,CIRERA A,MORANTE J R.Ab Initio Calculations of NO2and SO2Chemisorption onto Non-polar ZnO Surfaces[J].Sensors and Actuators B,2009,142:179.
[6]HU M,WANG W D,ZENG J,et al.Density Functional Theory Study of NO2-sensing Mechanisms of Pure and Ti-doped WO3(002)Surfaces[J].Chinese Physics B,2011,20(10):102101.
[7]HU M,ZHANG J,WANG W D,et al.Ab-Initio Density Functional Theory Study of a WO3NH3-sensing Mechanism [J].Chinese Physics B,2011,20(8):082101.
[8]BENJAWAN K,WARANYU P,BANCHOB W,et al.Density Functional Studies of Small Gases Adsorbed on the ZnO Sodalite-like Cage and Its Adsorption Abilities [J]. Computational and Theoretical Chemistry,2013,1020:100.
[9]MICHELLE J S S.Gas Sensing Applications of 1DNanostructured Zinc Oxide:Insights from Density Functional Theory Calculations[J].Progress in Materials Science,2012,57:437.
[10]JAVAD B,ALI A P,ZARGHAM B.Adsorption and Dissociation of Cl2Molecule on ZnO Nanocluster[J].Applied Surface Science,2012(258):8171.
[11]JAKUB S,JACEK PP,MICHAL L,et al.Density Functional Theory(DFT)Study of Zn,O2and O Adsorption on Polar ZnO(0001)and ZnO (0001)Surfaces[J].Journal of Crystal Growth,2013(374):53.
[12]BREEDON M,SPENCER M J S,YAROVSKY I.()Adsorption of NO and NO2on the ZnO 20 Surface:A DFT Study[J].Surface Science,2009,603:3389.
[13]LIU X,HU J,CHENG B,et al.First Principles Study of O2Adsorption on the LaFeO3(010)Surface[J].Sens.Actuators B ,2009,139(2):520.
[14]张德恒.透明导电膜中光吸收边的移动[J].半导体杂志,1998,23(3):34.ZHANG De-heng.Shift of Optical Absorption Edges in Transparent Conducting Films[J].Semiconductor,1998,23(3):34.(in Chinese)
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
云南化工(2021年8期)2021-12-21 06:37:16
原子与分子物理学报(2021年1期)2021-03-29 07:29:40
原子与分子物理学报(2021年1期)2021-03-29 07:28:18
陶瓷学报(2020年5期)2020-11-09 09:23:00
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
建材与装饰(2018年5期)2018-02-13 23:12:02
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
材料科学与工程学报(2016年1期)2017-01-15 13:34:08
贵州师范学院学报(2016年3期)2016-12-01 03:53:53
