
小白蛋白在新生大鼠缺氧后癫痫易感性升高中的作用机制
2014-08-11 08:25孙卫文詹丽璇
中风与神经疾病杂志 2014年3期
孙卫文,詹丽璇, 曾 玮,徐 恩
缺氧是导致新生儿缺氧性脑病和痫性发作的主要原因。新生儿期或围产期缺氧的患儿成年后对致痫因素的敏感性升高[1],其机制尚未明确。过度的Ca2+内流是导致细胞缺氧性损伤的关键因素。缺氧后,兴奋性神经元被激活,促进Ca2+内流,对细胞起毒性作用,抑制性神经元则反之。小白蛋白(parvalbumin,PV)是钙结合蛋白超家族中的一种,对Ca2+有很高的亲和力,能结合细胞内游离钙,缓冲Ca2+,对抗细胞内钙超载[2]和维持神经元网络的稳定[3]。本研究选用新生大鼠缺氧诱导痫性发作模型,观察缺氧后海马PV的表达,并检测海马Ca2+荧光强度,初步探讨PV在新生大鼠缺氧后癫痫易感性升高中的作用机制。
1 材料与方法
1.1 研究对象
新生10 d的Sprague-Dawley(SD)大鼠128只(由中山大学医学院实验动物中心提供),置于光照∶黑暗=12∶12标准光照周期的动物房喂养,母鼠与同窝新生大鼠同笼饲养。所有实验动物的操作及饲养均符合中华人民共和国实验动物管理条例。新生大鼠分为缺氧组和对照组,每组64只。
1.2 研究方法
1.2.1 制备缺氧性痫性发作模型 缺氧组采用改良的新生大鼠缺氧性痫性发作模型[4],大鼠置于恒温34℃的大小9000 cm2的密闭箱内,连续通入含有5%氧气、95%氮气的混和气体,对照组通入常氧(21%氧气、89%氮气的混和气体),共通气17 min。大鼠在进行缺氧时出现面部痉挛、节律点头、跌倒等表现。根据经典的Racine标准[2]对动物表现进行分级。出现全身性惊厥或大发作表现(即4、5级)的大鼠为入组标准。本研究中缺氧组大鼠缺氧时强直性阵挛发作表现较一致,重复性好,所有大鼠评分达到4、5级。
1.2.2 脑组织取材 分别于缺氧/常氧处理后1 d、3 d、7 d和14 d 2组各取16只大鼠取脑组织。麻醉后(用于流式细胞术检测的大鼠采用脊椎牵引脱臼法处死)于冰台上迅速分离海马组织,液氮快速冷冻后保存于-70℃冰箱中。另外,大鼠麻醉后用4℃生理盐水经心脏快速灌注,随后用4℃的4%多聚甲醛以先快后慢的速度固定,取脑经后固定、梯度蔗糖多聚甲醛溶液脱水、冰冻切片(片厚40 μm)后,选取相邻海马区脑片。各组中6只大鼠脑组织用于免疫组化,4只用于Western Blot,6只用于流式细胞术。
1.2.3 Nissl染色 1%焦油紫溶液染色15 min,梯度酒精脱水,二甲苯透明,封片。
1.2.4 免疫组化 脑片用0.01 mol/L的磷酸盐缓冲液(PBS,pH 值 7.4)洗 3 次各 5 min,3%H2O2室温孵30 min,加入稀释的鼠来源单克隆抗体PV(1∶8 000;Chemicon),4℃振荡孵育过夜。PBS洗3次,加入含生物素标记的羊抗鼠第二抗体,室温孵育2 h,PBS洗3次各5 min,加入含辣根过氧化物酶复合物,室温孵育30 min。PBS洗3次各5 min,DAB显色。梯度酒精脱水,透明,封片。
1.2.5 Western Blot 组织按 1 mg∶50 μl比例加入预冷的全细胞裂解液,匀浆后裂解30 min,20 000 r/min离心30 min(4℃)后提取上清液即为细胞总蛋白。BCA法进行蛋白定量。各组分别取等量的全细胞蛋白,用10%的 SDS聚丙烯酰胺(SDS-PAGE)进行电泳。100 V恒压转膜3 h。5%脱脂牛奶室温封闭1 h,TTBS漂洗10 min,3次,与一抗(PV 1∶8 000;β-actin 1∶1 000)4℃孵育过夜。二抗(羊抗鼠IgG 1∶1 000)室温反应1 h。ECL反应4 min后,暗室进行X-光胶片曝光、显影和定影。
1.2.6 流式细胞术 脑组织用0.25%胰蛋白酶消化30 min,以Dulbecco改良Eagle培养基(DuI-becco’s modified eagIe’s medium,DMEM)终止消化,过200目筛网,1000 r/min离心5 min。取细胞沉淀用Hanks液漂洗,离心5 min,将细胞沉淀用DMEM培养基制成密度为105~106细胞悬液,加入终浓度为5 μmol/L的fluo-3AM,置入37℃水浴箱中避光缓慢匀速50次/min振荡孵育30 min。1 000 r/min离心5 min,弃上清,Hanks液漂洗,如此2次。最后用含10%新生牛血清的DMEM制成密度105~106的细胞悬液。以X轴为荧光强度,Y轴为细胞数,以组方图检测样品的平均荧光强度值来表示神经元内[Ca2+]i的相对浓度大小。计算机存检数据,检测条件(包括负载时间、荧光剂强度、温度、电压等)保持恒定。
1.3 数据采集
在光学显微镜(×200)下,选取双侧海马CA1区、CA3区和DG区各3个连续视野,用Image J软件计数PV免疫阳性细胞数,每只大鼠共选取6张典型背侧海马脑片,计算其均数以精确实验数据。Western Blot X线胶片用Quantity One计算机图像分析系统分析,测定目标带灰度值,并以对照1 d组的灰度值为标准进行校正。
1.4 统计学方法
采用SPSS11.5进行统计分析。缺血组与对照组间的比较采用两因素(处理因素与时间因素)方差分析。进一步在同一处理组内不同时间点的比较用单因素方差分析,两两之间的分析采用Bonferreoni检验;相同时间点两组之间的比较采用独立样本的t检验。检验水准α为0.05。
2 结果
Nissl染色结果显示,缺氧组中海马各区细胞结构完整,胞浆内尼氏体无明显改变,未见细胞死亡。对照组海马各区细胞排列紧密,形态结构正常。
2.1 大鼠海马各区域PV免疫组化结果

图1 大鼠对照组(A~D)与缺氧组(E~H)海马PV免疫组织化学结果图
缺氧组和对照组海马各区均可见棕褐色PV免疫阳性细胞(见图1)。PV阳性神经元胞体及突起清晰可见,主要分布于海马CA1区的始层、CA3区的锥体细胞层和辐射层、齿状回的颗粒细胞层以及门区,可见有部分神经元的突起贯穿整个锥体细胞层。
各区域PV阳性细胞数如表1~表3所示。CA1区PV免疫阳性细胞数经两因素方差分析示,处理(缺氧与对照)主效应(F=53.87,P <0.01)、时间主效应(F=29.124,P <0.01)和两者交互效应(F=12.19,P <0.01)均有统计学意义。CA3区,处理主效应(F=38.50,P <0.01)、时间主效应(F=19.96,P <0.01)和两者交互效应(F=5.98,P <0.01)有统计学意义。
DG 区,处理主效应(F=56.28,P <0.01)、时间主效应(F=28.06,P <0.01)和两者交互效应(F=8.05,P <0.01)亦均有统计学意义。
缺氧组内,CA1区(F=5.69,P=0.02)、CA3区(F=6.28,P=0.02)、DG 区(F=3.41,P=0.04)各时间点PV免疫阳性细胞数有统计学差异,3 d和14 d均较1 d和7 d高(均P<0.05)。对照组内,各时间点 CA1区(F=53.87,P <0.01)、CA3区(F=38.50,P <0.01)、DG 区(F=40.50,P <0.01)PV 免疫阳性细胞数亦有统计学差异,3 d、7 d和14 d均较1 d高(均P<0.05)。与相应时间点对照组相比,在CA1区、CA3区和DG区,缺氧组PV免疫阳性细胞数7 d、14 d均降低(均 P <0.05),而缺氧1 d、3 d 与对照组比较差异无统计学意义(均P>0.05)。
2.2 大鼠海马PV蛋白的含量
如图2和表4所示:海马PV的含量经两因素方差分析示处理主效应(F=6.31,P<0.01)和时间主效应(F=8.71,P <0.01)有统计学意义,两者交互效应有统计学意义(F=14.02,P <0.01)。
缺氧组内,海马各时间点PV含量无统计学差异(F <0.01,P=0.98)。对照组内,各时间点 PV 含量有统计学差异(F=6.31,P <0.01),3 d、7 d 和 14 d均较1 d高(均P<0.05)。与相应时间点对照组相比,缺氧组3 d PV的含量增加,而7 d和14 d均降低(均 P <0.05)。
2.3 大鼠海马神经元内的Ca2+荧光强度
海马神经元内Ca2+荧光强度经两因素方差分析示,处理主效应(F=10.10,P <0.01)和时间主效应有统计学意义(F=10.19,P <0.01),两者交互效应有统计学意义(F=5.41,P=0.03)。缺氧组内,海马神经元内Ca2+荧光强度有统计学差异(F=9.23,P <0.01),3 d和7 d均较1 d降低(均 P <0.05)。对照组内,各时间点海马神经元内Ca2+荧光强度无统计学差异(F=2.23,P=0.1)。与相应时间点对照组相比,缺氧组海马神经元内Ca2+荧光强度均增加(均 P <0.01)(见表5)。

图2 各组海马PV Western Blot结果比较
表1 对照组与缺氧组大鼠海马CA1区PV免疫阳性细胞计数的比较()

表1 对照组与缺氧组大鼠海马CA1区PV免疫阳性细胞计数的比较()
与1 d比较,经Bonferroni法检验*P<0.05;与7 d比较,经Bonferroni法检验#P<0.05;与相同时间点的对照组比较,经独立样本t检验△P <0.05
1 d 3 d 7 d 14 d缺氧组对照组组别17.67 ±5.39 12.67 ±4.97 36.50 ±3.27*#31.33 ±6.38*19.17 ±3.813)32.83 ±6.94*34.00 ±3.69*#△44.00 ±3.29*
表2 对照组与缺氧组大鼠海马CA3区PV免疫阳性细胞计数的比较()

表2 对照组与缺氧组大鼠海马CA3区PV免疫阳性细胞计数的比较()
与1 d比较,经Bonferroni法检验*P<0.05;与7 d比较,经Bonferroni法检验#P<0.05;与相同时间点的对照组比较,经独立样本t检验△P <0.05
1 d 3 d 7 d 14 d缺氧组对照组组别14.83 ±5.35 13.50 ±3.78 30.00 ±6.69*#26.17 ±5.35*17.67 ±5.16△28.17 ±5.27*31.83 ±5.08*#△41.83 ±5.31*
表3 对照组与缺氧组大鼠海马DG区PV免疫阳性细胞计数的比较()
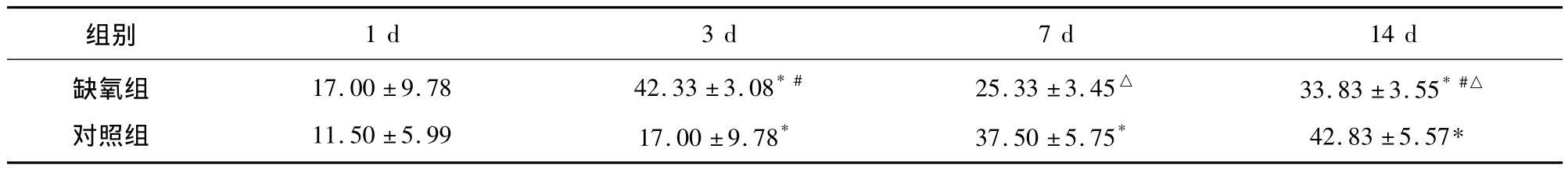
表3 对照组与缺氧组大鼠海马DG区PV免疫阳性细胞计数的比较()
1 d 3 d 7 d 14 d缺氧组对照组组别17.00 ±9.78 11.50 ±5.99 42.33 ±3.08*#17.00 ±9.78*25.33 ±3.45△37.50 ±5.75*33.83 ±3.55*#△42.83 ±5.57*
与1 d比较,经Bonferroni法检验*P<0.05;与7 d比较,经Bonferroni法检验#P<0.05;与相同时间点的对照组比较,经独立样本t检验△P <0.05
表4 缺氧组与对照组大鼠海马PV蛋白含量的比较()

表4 缺氧组与对照组大鼠海马PV蛋白含量的比较()
与1 d比较,经Bonferroni法检验*P<0.05;与相同时间点的对照组比较,经独立样本的t检验#P<0.05
1 d 3 d 7 d 14 d缺氧组对照组组别177.98 ±40.58 100.00 ±0.00 153.24 ±12.22#133.75 ±12.45*124.67 ±31.32#175.26 ±24.26*163.43 ±26.36#211.73 ±26.43*
表5 缺氧组与对照组大鼠海马神经元内Ca2+荧光强度的比较()

表5 缺氧组与对照组大鼠海马神经元内Ca2+荧光强度的比较()
与1 d比较,经Bonferroni法检验*P<0.05;与相同时间点的对照组比较,经独立样本的t检验#P<0.05
组别323.87 ±53.03#213.95 ±23.57 1 d 3 d 7 d 14 d缺氧组对照组362.57 ±92.78#210.17 ±74.12 192.03 ±36.93*#146.62 ±19.69 266.77 ±77.96*#163.97 ±48.89
3 讨论
缺氧造成的癫痫样发作多发生于儿童[5]。故新生大鼠缺氧诱导痫性发作模型是研究脑缺氧的理想方法之一。本研究通过建立新生大鼠缺氧诱导痫性发作模型,分别选择缺氧诱导痫性发作后不同的时间点,在国内外首次观察海马中 PV及神经元内Ca2+荧光强度的变化。
PV对 Ca2+有高度亲和力,能够缓冲细胞内Ca2+,保护神经元免受由于电压依赖性钙浓度过高而产生的损害,因而在中枢神经系统损伤后起着重要保护作用。本研究结果显示,在海马CA1区、CA3区及DG区缺氧后7~14 d PV阳性神经元表达减少,海马PV含量在缺氧后3 d较对照组增加,7~14 d则明显减少。提示缺氧诱导痫性发作后7~14 d在海马各区的PV神经元均受损。缺氧后3 d PV表达增加可能与缺氧后细胞内Ca2+迅速增加促使PV表达增加以加强对Ca2+的缓冲能力有关。Fagel等[6]对生后3~10 d的小鼠给予慢性缺氧,发现在缺氧后1 m皮质PV阳性神经元较正常减少;Wittner等[7]的研究发现,在颞叶癫痫患者的海马非硬化CA1区PV在胞体的表达减少,严重海马硬化的癫痫患者海马CA1区仅有少量 PV阳性细胞表达。Schwaller等[8]的研究发现,在戊四氮诱导痫性发作模型中,PV-/-基因型动物癫痫发作程度要比PV+/+严重,推测由于PV神经元功能或数量的减少导致大鼠癫痫易感性升高。结合我们前期研究结果[9],推测PV神经元的损伤及含量的减少是γ-氨基丁酸在缺氧导致新生大鼠癫痫易感性升高中起作用的机制之一。其机制可能与对细胞内Ca2+缓冲能力减弱、神经元兴奋性增高及影响γ-氨基丁酸能中间神经元的快速放电能力,减弱对锥体细胞的抑制性有关。
缺氧后细胞内外离子稳态失衡造成细胞死亡。Silver等[10]的研究发现,在正常情况下神经元内Ca2+浓度为 10-8~10-7mol/L,且各脑区间无明显差别,然而在缺氧或血流阻断30~60 s后,在皮质观察到微小的细胞内Ca2+增加,而海马CA1区神经元内Ca2+浓度可明显增加至6×10-4~8×10-4mol/L。研究已证明,癫痫灶脑细胞内Ca2+浓度升高达阈限以上是癫痫发生和癫痫脑损害的直接原因。本研究显示:与各时间点的对照组相比,缺氧组海马神经元内Ca2+荧光强度明显增高,且缺氧后1 d海马神经元内Ca2+荧光强度高于缺氧后3 d及7 d,提示缺氧后海马神经元内Ca2+浓度迅速增加,我们推测由于PV神经元发育尚不完全,因此缺氧后1 d海马神经元内Ca2+浓度明显增高,并且刺激海马PV神经元的表达以适应神经元内Ca2+浓度的增加,随着PV神经元的发育及代偿性增加,其对Ca2+的缓冲能力增加,细胞内Ca2+浓度逐渐下降,但是在缺氧后7 d时,PV神经元明显减少,对抗Ca2+超载能力减弱,不能有效地缓冲细胞内Ca2+导致缺氧后14 d时出现细胞内Ca2+浓度较缺氧后3 d升高。因此我们认为缺氧对其所致的Ca2+超载导致神经元兴奋性增加,是引起癫痫发作的机制之一。由于实验条件及动物模型的限制,本研究未能对海马进行分区检测神经元内Ca2+荧光强度。
综上所述,我们推测新生大鼠缺氧诱导痫性发作后PV神经元的损伤、对细胞内游离Ca2+缓冲能力减弱、影响中间神经元的快速放电能力,减弱对锥体细胞的抑制,最终导致海马神经元兴奋性增加是PV在新生大鼠缺氧诱导痫性发作后癫痫易感性升高中起作用的机制之一。缺氧后细胞内游离Ca2+增加可能是导致PV神经元损伤的原因之一。
[1]Rakhade SN,Klein PM,Huynh T,et al.Development of later life spontaneous seizures in a rodent model of hypoxia-induced neonatal seizures[J].Epilepsia,2011,52(4):753 -765.
[2]Pauls TL,Cox JA,Berchtold MW.The Ca2+-binding proteins parvalbumin and oncomodulin and their genes:new structural and functional findings[J].Biochimicaet Biophysica Acta,1996,1306(1):39 -54.
[3]Fuchs EC,Zivkovic AR,Cunningham MO,et al.Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior[J].Neuron,2007,53(4):591 -604.
[4]Racine RJ.Modification of seizure activity by electrical stimulation.II.Motor seizure[J].Electroencephalogr Clin Neurophysiol,1972,32(3):281-294.
[5]Sankar MJ,Agarwal R,Aggarwal R,et al.Seizures in the newborn[J].Indian J Pediatr,2008,75(2):149 -155.
[6]Fagel DM,Ganat Y,Cheng E,et al.Fgfr1 is required for cortical regeneration and repair after perinatal hypoxia[J].J Neurosci,2009,29(4):1202-1211.
[7]Wittner L,Eross L,Czirják S,et al.Surviving CA1pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus[J].Brain,2005,128(Pt1):138 -152.
[8]Schwaller B,Tetko IV,Tandon P,et al.Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures[J].Mol Cell Neurosci,2004,25(4):650 -663.
[9]孙卫文,詹丽璇,李 珂,等.γ-氨基丁酸能神经元在缺氧性痫性发作中的作用[J].中风与神经疾病杂志,2013,30(2):132-136.
[10]Silver IA,Erecinska M.Intracellular and extracellular changes of[Ca2+]in hypoxia and ischemia in rat brain in vivo[J].J Gen Physiol,1990,95(5):837 - 866.
猜你喜欢
中国现代医生(2022年23期)2022-09-21
作文周刊·小学二年级版(2022年20期)2022-05-05
中华养生保健(2020年2期)2020-11-16
中国病理生理杂志(2020年3期)2020-04-03
野生动物学报(2020年1期)2020-02-21
创新作文(小学版)(2019年10期)2019-09-25
饮食科学(2017年5期)2017-05-20
中国高原医学与生物学杂志(2017年4期)2017-03-08
安徽医科大学学报(2015年9期)2015-12-16
汽车观察(2009年1期)2009-02-18
