
16p11.2缺失综合征1例并文献复习
2014-08-10 12:28肖咏梅陆燕芬张育才
中国循证儿科杂志 2014年6期
葛 婷 崔 云 肖咏梅 陆燕芬 张育才 张 婷
·论著·
16p11.2缺失综合征1例并文献复习
葛 婷1崔 云1肖咏梅 陆燕芬 张育才 张 婷

16号染色体; 16p11.2缺失综合征; 脊柱侧弯
1 病例资料
男,2月13 d,因“发热近20 d伴咳嗽、腹泻”入住上海市儿童医院(我院)。父母非近亲婚配,母孕期间和出生史未述不良事件,否认家族遗传疾病史。
图1为患儿起病后的症状、体征、诊断和治疗等重要临床信息时间轴。
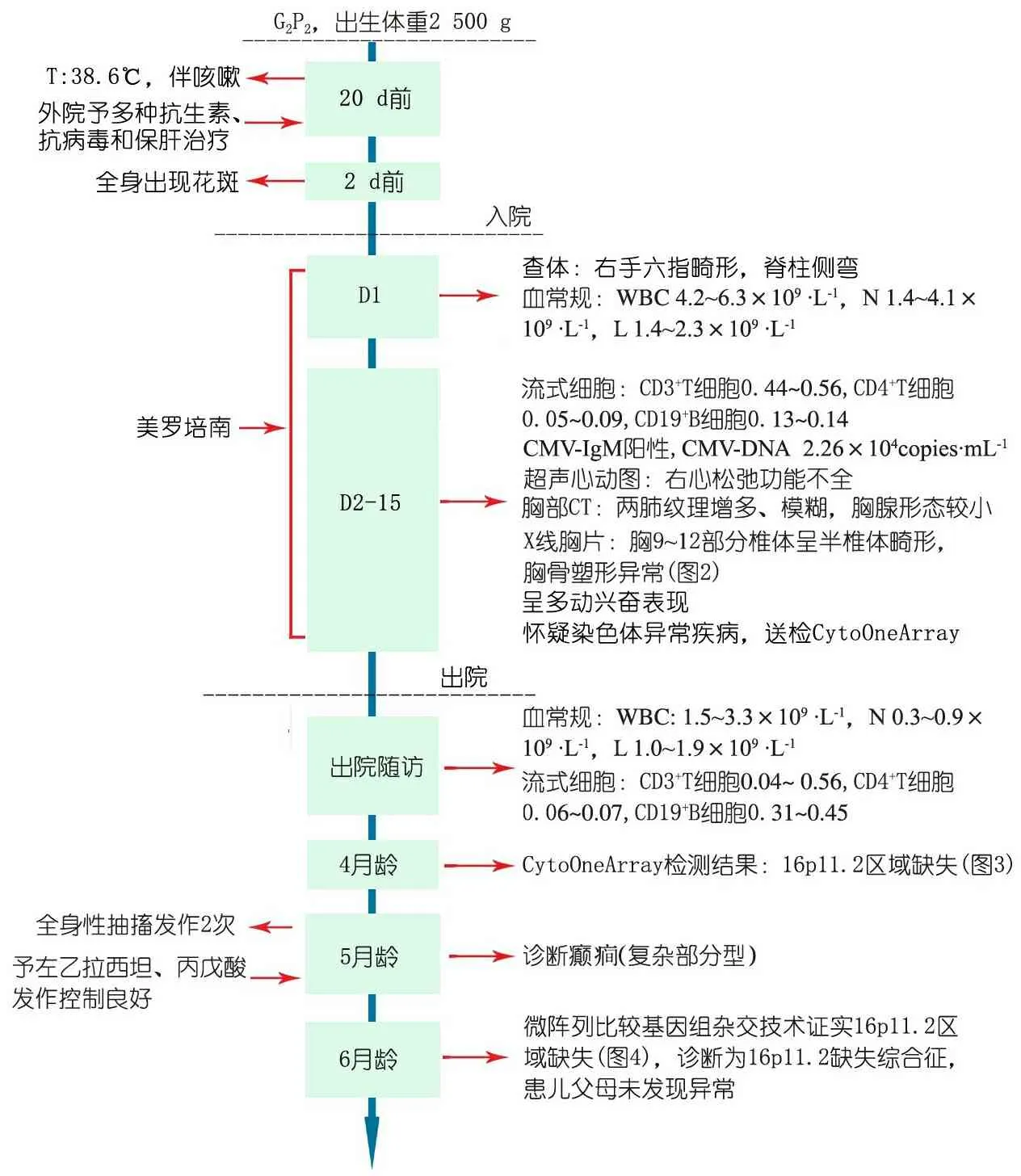
图1 本文病例重要临床信息时间轴
Fig 1 Time shaft of important clinical information of the patient in the study
患儿入我院前2 d头面部及双上肢出现少许出血点,伴纳差、腹泻、精神状态不佳。
入院查体:T:38.2℃,P 140·min-1,R 40·min-1,BP 76/54 mmHg,神清,气促,反应一般,双侧瞳孔等大等圆,对光反射灵敏,全身花纹样改变,压之褪色,双肺呼吸音粗,可闻及少许痰鸣音,心音有力,律齐,未闻及明显杂音,腹软不胀,肝肋下2cm,质软,脾肋下未及,四肢肌力肌张力正常。

图2 本文病例脊柱侧弯及半椎体畸形
Fig 2Scoliosis and half vertebral deformity of the 9thto 12thvertebral body in the case

图3 CytoOneArray检测16号染色体结果
Fig 3Results of chromosome 16 detected by GytoOneArray

图4 CGH Microarray验证Chr 16p11.2缺失区段结果
Fig 4 16p11.2 deletion checked by CGH microarray
2 讨论
染色体病是由于先天性染色体数目异常或结构异常而引起的临床综合征。中国新生儿出生缺陷的发病率为4%~6%, 其中大部分为染色体异常所致,其中染色体部分缺失的发生率较低,但种类繁多,症状缺乏特异性,患儿多于婴幼儿期夭折。
16p11.2缺失综合征指涉及16号染色体短臂1区1带2亚带上不同大小片段的缺失,该区域不同程度的缺失所致的临床表现复杂多样,可影响机体认知功能、行为、生长发育和BMI等[1~5]。以“16p11.2[All Fields] AND (“sequence deletion”[MeSH Terms] OR (sequence[All Fields] AND deletion[All Fields]) OR “sequence deletion”[All Fields] OR deletion[All Fields])”为检索式检索PubMed数据库,以检索词“16p.11.2综合征”检索中国万方数据库和中国知网,共复习16p.11.2综合征1 378例,临床表型涉及到神经系统表现547例(39.7%)、内分泌系统371例(26.9%)、生长发育与骨骼异常84例(6.1%)、泌尿生殖与消化系统10例(0.7%)、心血管系统4例(0.3%)、免疫功能异常1例(0.07%)。
16p11.2区域中涉及可调控瘦素和胰岛素信号的SH2B1基因,已有报道涵盖该基因片段的16p11.2缺失综合征患儿中,0.5%患有严重的早发性肥胖,主要表现为极端的食欲过盛,严重的胰岛素抵抗,最终可致生长发育迟缓[6~9]。16p11.2上基因片段的缺失和重复也是孤独症(ASD)和神经发育障碍最常见的致病基因之一,这可能与该区域内涵盖了编码人MAPK3基因,智力低下相关基因(ALOX5、ACSL4、PTGS2、HPRT1) ,ASD相关基因(MVP、CDIPT1、SEZ6L2、ASPHD1、KCTD13)有关,从而导致部分患儿出现智力低下、自闭的神经精神发育异常,但总人群发生率<0.01%[10~16]。
Nik-Zainal等[17]研究证实63例苗勒管发育不全患者中,4例为16p11.2区域内涉及约0.55 Mb基因微缺失造成。16p11.2缺失综合征与多发畸形有关,以骨骼多发畸形居多,包括头围异常、身材矮小、虹膜缺损、小眼畸形、椎骨和脊柱的畸形,还包括脊髓空洞症,先天性膈疝[4, 16,18~25]。有研究报道位于16p11.2的TBX6基因的多态性与汉族人群的脊柱侧弯相关[26]。本文病例胸部X线可见胸9~12椎体部分呈半椎体畸形,胸骨塑形异常(脊柱侧弯),符合16p11.2缺失综合征的表型。
由于16p11.2缺失综合征患儿缺失片段大小不一,因此该类患儿临床表型具有较大的异质性,临床表型可为智力障碍、先天发育异常、ASD、语言障碍,也可为正常表现[35]。目前针对16p11.2缺失综合征尚无有效的治疗方法,遗传咨询和产前检查能降低染色体缺陷患儿的出生,本文患儿其远期生存情况,精神运动发育和骨骼畸形矫正等尚待进一步追踪随访。
[1]Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature, 2010, 463(7281): 671-675
[2]Dale RC, Grattan-Smith P, Fung VS, et al. Infantile convulsions and paroxysmal kinesigenic dyskinesia with 16p11.2 microdeletion. Neurology, 2011, 77(14): 1401-1402
[3]Zufferey F, Sherr EH, Beckmann ND, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet, 2012, 49(10): 660-668
[4]Mefford HC, Cooper GM, Zerr T, et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res, 2009, 19(9): 1579-1585
[5]Bochukova EG, Huang N, Keogh J, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature, 2010, 463(7281): 666-670
[6]Perrone L, Marzuillo P, Grandone A, et al. Chromosome 16p11.2 deletions: another piece in the genetic puzzle of childhood obesity. Ital J Pediatr, 2010, 3643
[7]Bachmann-Gagescu R, Mefford HC, Cowan C, et al. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet Med, 2010, 12(10): 641-647
[8]Galioto R, Spitznagel MB, Strain G, et al. Cognitive function in morbidly obese individuals with and without binge eating disorder. Compr Psychiatry,2012, 53(5): 490-495
[9]Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature, 2011, 478(7367): 97-102
[10]Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet, 2011, 43(12): 1252-1255
[11]Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med, 2008, 358(7): 667-675
[12]McCarthy SE, Makarov V, Kirov G, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet, 2009, 41(11): 1223-1227
[13]Crepel A, Steyaert J, De la Marche W, et al. Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am J Med Genet,2011,156(2): 243-245
[14]Kumar RA, KaraMohamed S , Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Molec Genet, 2008,17(4):628-638
[15]Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet, 2008, 82(2): 477-488
[16]Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant.Nature,2012,485(7398):363-367
[17]Nik-Zainal S, Strick R, Storer M, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J Med Genet, 2011, 48(3): 197-204
[18]Finelli P, Natacci F, Bonati MT, et al. FISH characterisation of an identical (16)(p11.2p12.2) tandem duplication in two unrelated patients with autistic behaviour. J Med Genet, 2004,41(7): e90
[19]Bourthoumieu S, Esclaire F, Terro F, et al. First prenatally diagnosed case of 16p11.2p12.1 duplication. Prenat Diagn, 2008, 28(3): 254-256
[20]Bedoyan JK, Kumar RA, Sudi J, et al. Duplication 16p11.2 in a child with infantile seizure disorder. Am J Med Genet A, 2010, 152A(6): 1567-1574
[21]Shinawi M, Liu P, Kang SH,et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet, 2010,47(5): 332-341
[22]Hernando C, Plaja A, Rigola MA, et al. Comparative genomic hybridisation shows a partial de novo deletion 16p11.2 in a neonate with multiple congenital malformations. J Med Genet,2002, 39(5): E24
[23]Schaaf CP, Goin-Kochel RP, Nowell KP, et al. Expanding the clinical spectrum of the 16p11.2 chromosomal rearrangements: three patients with syringomyelia. Europ J Hum Genet, 2011,19(2): 152-156
[24]Shimojima K, Inoue T, Fujii Y, et al. A familial 593-kb microdeletion of 16p11.2 associated with mental retardation and hemivertebrae. Eur J Med Genet, 2009, 52(6): 433-435
[25]Wat MJ, Veenma D, Hogue J, et al. Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic hernia. J Med Genet,2011,48(5): 299-307
[26]Fei Q, Wu Z, Wang H, et al. The association analysis of TBX6 polymorphism with susceptibility to congenital scoliosis in a Chinese Han population. Spine (Phila Pa 1976), 2010,35(9): 983-988
[27]Shiow LR, Paris K, Akana MC, et al. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol, 2009, 131(1): 24-30
[28]Shiow LR, Roadcap DW, Paris K, et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immun, 2008, 9(11): 1307-1315
[29]Weber A, Köhler A, Hahn A, et al. Benign infantile convulsions (IC) and subsequent paroxysmal kinesigenic dyskinesia (PKD) in a patient with 16p11.2 microdeletion syndrome. Neurogenetics, 2013, 14(3-4): 251-253
[30] Castro-Gago M, Pérez-Gay L, Gómez-Lado C, et al.16p11.2 microdeletion associated to early onset benign childhood seizures. Rev Neurol, 2013, 56(2): 125-127
[31] Dale RC, Grattan-Smith P, Fung VS, et al.Infantile convulsions and paroxysmal kinesigenic dyskinesia with 16p11.2 microdeletion. Neurology, 2011,77(14): 1401-1402
[32] Battaglia A, Novelli A, Bernardini L, et al. Further characterization of the new microdeletion syndrome of 16p11.2-p12.2. Am J Med Genet A, 2009,149A(6): 1200-1204
[33]Tabet AC, Pilorge M, Delorme R, et al. Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother. Eur J Hum Genet, 2012,20(5): 540-546
[34]Shen Y, Chen X, Wang L, et al. Intra-family phenotypic heterogeneity of 16p11.2 deletion carriers in a three-generation Chinese family. Am J Med Genet B Neuropsychiatr Genet, 2011, 156(2): 225-232
[35]Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet, 2009, 52(2-3): 77-87
(本文编辑:丁俊杰)
One case of chromosome 16p11.2 deletion syndrome and literature review
GETing1,CUIYun1,XIAOYong-mei,LUYan-fen,ZHANGYu-cai,ZHANGTing
(ShanghaiChildren′sHospital,ShanghaiJiaoTongUniversity,Shanghai200062,China;1Equalcontributiontothisstudy)
Corresponding Author:ZHANG Ting,E-mail:zhangt@shchildren.com.cn; ZHANG Yu-cai,E-mail:Zhangyc@shchildren.com.cn
ObjectiveTo enhance the understanding of clinical characteristics,diagnosis,follow-up and genetic testing of chromosome 16p11.2 deletion syndrome.Methods The clinical manifestations,laboratory testing,diagnosis,follow-up,and genetic testing of one case with chromosome 16p11.2 deletion syndrome were reviewed,analyzed and summarized. Meanwhile, relevant literatures of chromosome 16p11.2 deletion syndrome were reviewed in this article.Results①A 2-month-and-13-day boy with 20-day fever,cough,and diarrhea was admitted to our hospital. Deformity of six fingers in right palm and scoliosis was found. The total peripheral blood lymphocytes and lymphocyte subsets were lower than the reference levels. Chest X-ray indicated that the sternum shape was abnormal and T9-T12 vertebral bodies were hemivertebrae deformity. The patient was improved with a hyperactive and exciting performance after anti-infection therapy. Follow up after releasing indicated that the count of peripheral blood lymphocytes was improved, however, WBC, N and CD4+T cells remained low levels. The boy was diagnosed as epilepsy at 5 months old and improved after treatment with anti-epileptic drugs. A deletion of 0.545 4 Mb in chromosome 16p11.2 was identified by chromosome chip detection technology and confirmed by high-density oligonucleotide comparative genomic hybridization (CGH) Microarray. The genes located in this deleted region includedSPN,QRRT,C16orf54,KIF22,MAZ,SEZ6L2,CDIPT,ASPHD1,KCTD13,TMEM219,TAOK2,DOC2A,TBX6. The results of Chromosome chip detection were normal in his parents. Thus, this boy was finally diagnosed as chromosome 16p11.2 deletion syndrome. ②1 387cases were reported by 95 published articles related with chromosome 16p11.2 deletion syndrome, involving the nervous system(547,39.7%), endocrine system(371,26.9%), growth and skeletal abnormalities(84,6.1%), urinary and digestive system(10,0.7%),cardiovascular system(4,0.3%), immune function(1,0.07%). The different size of the deletion region in chromosome 16p11.2 led a high heterogeneity of clinical characteristics.ConclusionChromosome 16p11.2 deletion syndrome has variable clinical manifestations ,including multiple skeletal deformities(such as scoliosis), nervous system abnormalities(such as seizure, autism), other systems(such as repeated infection, endocrine abnormalities). The diagnosis of Chromosome 16p11.2 deletion syndrome relies on chromosome chip detection technology and CGH microarray.
Chromosome 16; Chromosome 16p11.2 deletion syndrome; Gene expression; Scoliosis
上海市儿童医院,上海交通大学附属儿童医院 上海,200062,1 共同第一作者
张婷,E-mail:zhangt@shchildren.com.cn; 张育才,E-mail:Zhangyc@shchildren.com.cn
10.3969/j.issn.1673-5501.2014.06.010
2014-08-20
2014-12-02)
猜你喜欢
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
中国生殖健康(2020年6期)2020-02-01
中国循证儿科杂志(2019年1期)2019-03-29
科学之谜(2019年3期)2019-03-28
河北农业科学(2019年6期)2019-03-21
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中南医学科学杂志(2015年4期)2015-12-28
