
硫代磷酸化修饰的反义TGF-β1寡核苷酸抑制血管损伤后内膜增殖*
2014-08-09 00:41林志鸿谢良地吴可贵李庚山蔺佩鸿
中国病理生理杂志 2014年8期
林志鸿, 谢良地, 吴可贵, 李庚山, 蔺佩鸿
(福建医科大学 1附属第一医院急诊科, 2附属第一医院干部病房, 3福建省高血压研究所,福建 福州 350005; 4武汉大学人民医院心内科,湖北 武汉 430000)
血管损伤后血管平滑肌细胞(vascular smooth muscle cells,VSMCs)的过度增殖及随后的细胞外基质(extracellular matrix, ECM)沉积在血管损伤后的再狭窄过程中扮演着至关重要的作用。血管损伤后,VSMCs许多特征与高血压时的VSMCs相似,表现为明显的生长合成表型、更快的生长速度、异常的生长接触抑制及加速进入S期的细胞生长周期。同时对许多生长因子呈现出非特异性的高增殖倾向[1]。既往研究表明,血管损伤后转化生长因子β1(transforming growth factor β1,TGF-β1)的表达水平明显升高,且TGF-β1显著促进了血管损伤后VSMCs的异常增殖,促进了ECM的合成及在损伤血管内膜的堆积[2-3]。因此抑制损伤血管局部TGF-β1的高表达,将可能显著抑制VSMCs的异常增殖及ECM在损伤血管内膜的堆积,从而减轻血管损伤后内膜的增殖和肥厚。
材 料 和 方 法
1 反义寡核苷酸的设计及合成
应用Wilbar-Lipman方法,寻找出大鼠TGF-β1的cDNA序列[4],在跨过cDNA序列的起始密码区ATG范围内,设计包含有15个碱基、无修饰或硫代磷酸化修饰的反义TGF-β1寡核苷酸,同时设计与此反义寡核苷酸相对应的、作为对照的正义寡核苷酸(与反义寡核苷酸互补),无修饰的寡核苷酸应用于离体的细胞培养实验,而硫代磷酸化修饰的寡核苷酸则用于在体治疗实验。具体如下:反义 TGF-β1(AS TGF-β1): 5’-CGAGGGCGGCATGGG-3’;正义TGF-β1(S TGF-β1): 5’-GCTCCCGCCGTACCC-3’。上述所有的寡核苷酸均采用Model 394 DNA Synthesizer (Applied Biosystems)合成,应用OPC column(Applied Biosystems)纯化。
2 Sprague-Dawley(SD)大鼠颈总动脉内膜球囊损伤
体重约400~500 g的雄性SD大鼠,由福建医科大学实验动物中心提供,氯氨酮(40 mg/kg)麻醉,无菌操作暴露右颈总动脉,分离至动脉交叉处,结扎颈外动脉远端及其分支,同时暂时阻断颈总动脉近心端及颈内动脉血流。在离动脉交叉处远心端约0.5~1 cm处切开颈外动脉,插入2F的球囊导管(Baxter Healthcare)约2.0 cm,从导管注入0.08 mL生理盐水扩张球囊,有明显阻力感后来回拖拉3次,确定造成血管壁的损伤后退出球囊导管,在靠近动脉交叉处结扎颈外动脉,同时开放颈内及颈总动脉使血流再通后,缝合切口。
3 SD大鼠正常及损伤颈总动脉VSMCs的培养
上述大鼠血管球囊损伤术后1周无菌操作取出正常或损伤的颈总动脉约1~1.5 cm,置入含RPMI-1640基础培养液(Gibco)的无菌培养皿中,采用组织块翻转干固法,将组织块均匀贴附于30 mL的螺口培养瓶。从第3天起,用倒置相差显微镜(Olympus)观察细胞生长情况,应用alpha-smooth muscle actin单克隆抗体(DAKO)进行免疫细胞荧光鉴定。用3~5代细胞进行实验。
4 VSMCs增殖([3H]-TdR掺入率)的检测
传代培养的VSMCs进行反义寡核苷酸的干预实验。将反义及正义的TGF-β1用无血清的RPMI-1640培养基稀释后,加入处于同步化状态的无血清的VSMCs,使干预寡核苷酸的最终浓度分别为0、0.01、0.1和1 μmol/L。在干预药物加入时,同时加入3.7×107Bq/L[3H]-胸腺嘧啶核苷酸([3H]-thymidine,[3H]-TdR;中国原子能科学研究院),共育24 h后,0.25%胰酶消化,在0.45 μm微孔滤膜上负压抽滤,生理盐水和10%三氯醋酸冲洗滤膜,室温凉干后置入闪烁杯中,加入2,5-二苯基噁唑/1,4-双[2-(5-苯基)噁唑基)苯(PPO/POPOP)/二甲苯闪烁液4 mL,静置过夜后,在液体闪烁计数器(Tri-Carb 2300;PerkinElmer) 上进行放射性强度的测定。
5 RT-PCR检测VSMCs TGF-β1 、纤维连结蛋白(fibronectin, FN) 及平滑肌22α蛋白(smooth muscle 22α, SM22α)、基质Gla蛋白(matrix Gla)及骨桥蛋白(osteopontin)mRNA的表达
将1×108cells/L VSMs接种于24孔板,每孔1 mL,培养24 h贴壁后,换用无血清的RPMI-1640培养基,继续培养24 h,使细胞处于同步化状态。加入AS TGF-β1干预24 h后,提取细胞总RNA,反转录为cDNA后进行PCR扩增 (TaKaRa Biochemicals)。各引物序列根据文献报道及应用Primer 3软件设计合成。采用18S rRNA作为内参照,进行产物电泳条带强度的半定量分析。引物序列及产物长度见表1。

表1 引物序列
6 Western blotting检测VSMCs TGF-β1蛋白的表达
将4~6代VSMCs接种于6孔板贴壁后,换用无血清的RPMI-1640继续培养24 h,细胞同步化后加入AS TGF-β1及S TGF-β1。干预24 h后提取细胞总蛋白质,煮沸变性10 min,按每孔5 μg蛋白质及20 μL样本缓冲液,在8% SDS-PAGE上样电泳约3 h,后转至硝酸纤维素膜上,应用TGF-β1单克隆抗体(Santa Cruz)检测蛋白质的表达,以α-tubulin(Sigma)作为内参照。
7 SD大鼠颈动脉球囊导管损伤及AS TGF-β1的干预治疗
将雄性SD大鼠分为4组,每组4只,血管内膜球囊导管损伤方法同前及本实验室以前发表的文章[5]。第1组为假手术组(仅进行颈动脉的暴露和分离,没有血管球囊损伤),第2组为手术损伤血管后仅给予生理盐水2.5 μL/h, 第3组为手术损伤后给予硫代磷酸化修饰的S TGF-β1(90 g·kg-1·d-1),最后1组为手术损伤后给予硫代磷酸化修饰的AS TGF-β1(90 μg·kg-1·d-1)。所有给药途径均采用ALZET 泵从皮下注射,连续给药28 d,每7 d根据体重的变化调整药物用量。术后28 d迅速取出损伤或未损伤的颈总动脉约1 cm,去除血液及周围结缔组织后置于4%甲醛中,常规脱水、石蜡包埋,切成6~10 μm的薄片,HE染色。应用图像分析仪(CMIAS-B型多功能真彩色病理图像分析系统,北京航空航天大学图像中心)摄像求积法测定血管的内膜与中膜的面积比(intima/media,I/M)。
8 统计学处理
采用SPSS 13.0软件分析。数据以均数±标准差(mean±SD)表示。组间差异比较用单因素方差分析(One-way ANOVA),两两比较采用Student-Newman-Keuls检验。以P<0.05为差异有统计学意义。
结 果
1 AS TGF-β1对血管损伤后VSMCs TGF-β1 mRNA表达的影响
各种浓度(0.01~1 μmol/L)的AS TGF-β1对血管损伤后VSMCs TGF-β1mRNA表达并无明显作用,表明AS TGF-β1的作用环节并不在转录水平上,因为它并不影响TGF-β1mRNA的生成,见图1。
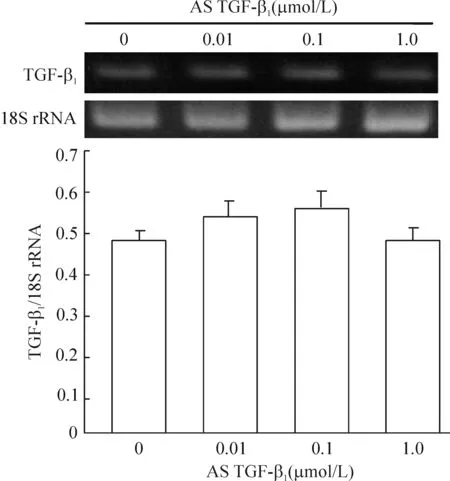
Figure 1. Effects of AS TGF-β1 on the expression of TGF-β1 mRNA in VSMCs after vascular injury. Mean±SD. n=4.
2 AS TGF-β1对血管损伤后VSMCs TGF-β1蛋白表达的影响
Western blotting检测结果表明,AS TGF-β1呈浓度依赖性地抑制损伤VSMCs TGF-β1蛋白的表达(P<0.05),而S TGF-β1对损伤后VSMCs过度表达的TGF-β1蛋白并无抑制作用(P>0.05)。这表明AS TGF-β1的作用机制是抑制了TGF-β1mRNA的翻译过程,从而抑制了TGF-β1蛋白在损伤VSMCs的过度表达,见图2。
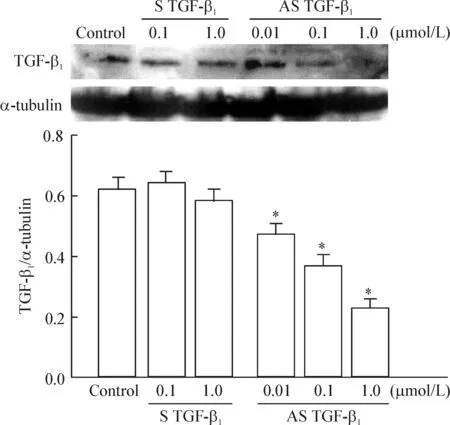
Figure 2. Effects of S TGF-β1 and AS TGF-β1 on the expression of TGF-β1 protein in VSMCs after vascular injury. Mean±SD. n=4. *P<0.05 vs control group.
3 AS TGF-β1对血管损伤后VSMCs 增殖的影响
AS TGF-β1呈浓度依赖性地抑制血管损伤后VSMCs DNA的合成,S TGF-β1对VSMCs [3H]-TdR的掺入率并无明显作用;不管是反义、还是正义的TGF-β1寡核苷酸对非损伤血管的VSMCs DNA的合成均无明显的量效作用,见图3。
4 AS TGF-β1对血管损伤后VSMCs合成ECM的影响
AS TGF-β1显著抑制了血管损伤后VSMCs FN的合成,且AS TGF-β1的抑制作用呈浓度依赖性,见图4。表明血管损伤后VSMCs合成ECM增多,而AS TGF-β1有抑制VSMCs合成和分泌ECM的作用,从而降低血管损伤后ECM的沉积,抑制血管新生内膜的形成。
5 AS TGF-β1对血管损伤后VSMCs表型的作用
AS TGF-β1对代表收缩表型的SM22α mRNA的表达水平呈现明显的促进作用;而对合成表型标志物的基质Gla蛋白和骨桥蛋白mRNA表达水平则呈现显著的抑制作用,这两种相反的作用均在0.01及0.1 μmol/L剂量最为明显,见图5。
6 AS TGF-β1及S TGF-β1对血管损伤后新生内膜形成的作用
血管损伤后,内膜明显增厚,管腔狭小,而中膜并无显著变化,I/M较血管未损伤组明显增加(0.19±0.03vs1.76±0.16,P<0.01);AS TGF-β1治疗4周显著抑制了血管损伤后新生内膜的形成,增加了管腔面积,降低了I/M,为损伤组的32%(1.76±0.16vs0.56±0.08,P<0.01);S TGF-β1对血管损伤后新生内膜的肥厚并无显著影响,I/M与损伤组无明显差别(1.55±0.11vs1.76±0.16,P>0.05),见图6。

Figure 3. Effects of S TGF-β1 and AS TGF-β1 on the DNA synthesis in VSMCs with (A)or wihtout (B) vascular injury. Mean±SD. n=4. *P<0.05 vs 0 μmol/L.
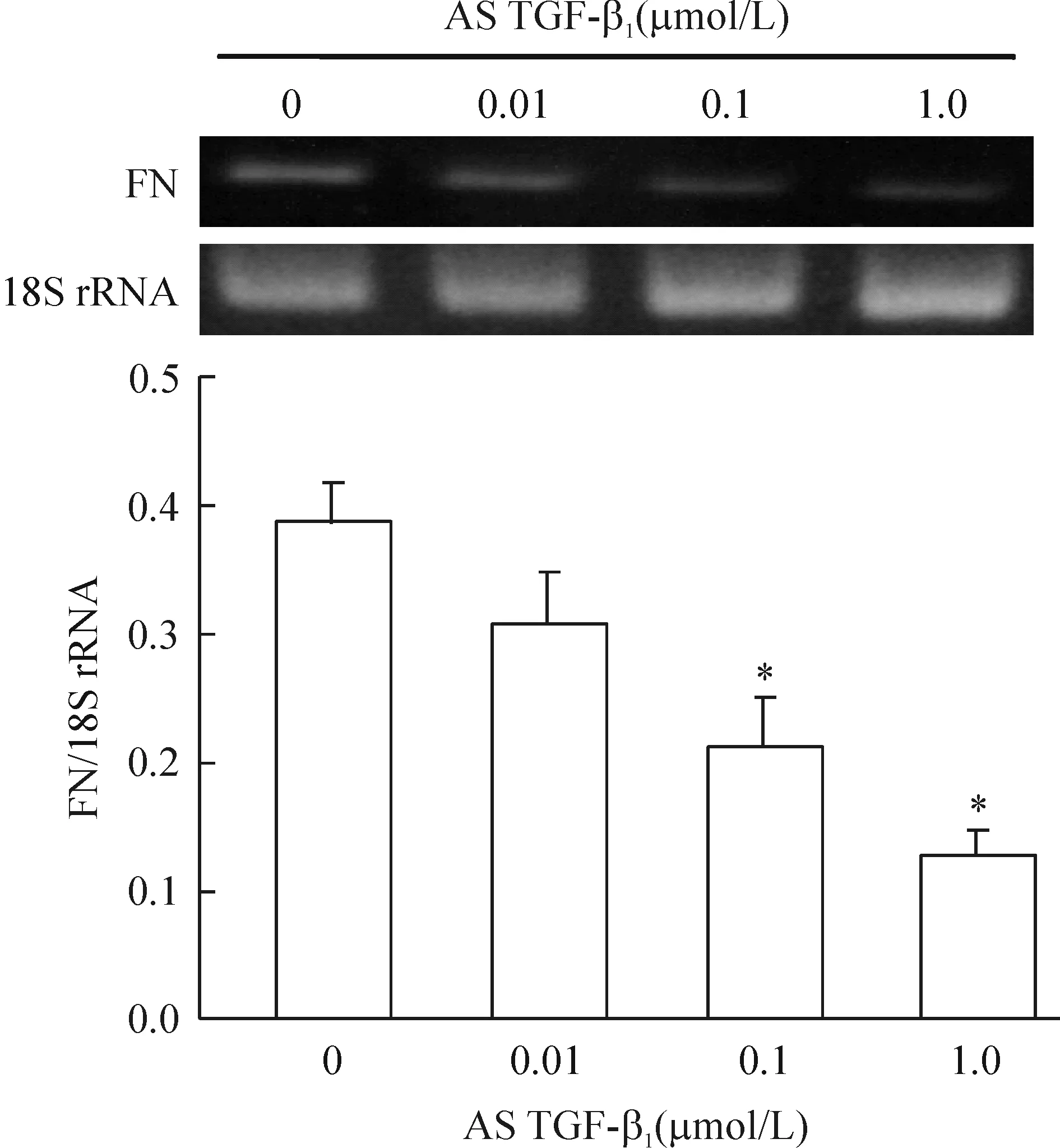
Figure 4. Effects of AS TGF-β1 on the expression of FN mRNA in VSMCs after vascular injury. Mean±SD. n=4. *P<0.05 vs 0 μmol/L.
讨 论
反义寡核苷酸是一种未修饰或经过化学修饰的单链DNA分子,一般由13~25个核苷酸组成,通过细胞内化作用,短寡核苷酸可进入细胞内,进入胞内的反义寡核苷酸与其目标mRNA特异性结合,阻断了目的mRNA的翻译,从而抑制该蛋白质表达,影响了细胞相应的功能[6]。这种特异性的结合,使得反义寡核苷酸技术在有选择性地调整和改变某些与疾病病理过程有关的基因方面,变得越来越具有吸引力。
我们的研究结果也证实AS TGF-β1呈浓度依赖性抑制了血管损伤后VSMCs TGF-β1蛋白质的过度表达,但并不影响转录水平上TGF-β1mRNA的生成,而S TGF-β1并不影响TGF-β1蛋白质的表达。表明我们所设计的反义TGF-β1寡核苷酸能够特异性地制VSMC TGF-β1的表达水平,而且这种作用主要表现在翻译水平上。
已有研究发现,损伤血管内膜TGF-β1的表达明显升高,且显著促进了损伤血管VSMCs的异常增殖、ECM的合成及在损伤血管内膜的堆积[7]。本研究中,我们发现AS TGF-β1呈浓度依赖性地抑制了血管损伤后VSMCs DNA的合成,减少VSMCs合成和分泌ECM-FN,但对未损伤血管的VSMCs并无明显作用。在血管损伤后初期对VSMCs的抑制作用,也许足于减少血管壁ECM的堆积,及至抑制血管损伤后内膜肥厚的最终形成。有研究表明在损伤后2周,TGF-β1mRNA的表达水平可达到正常的5~7倍,因此若能在损伤早期抑制TGF-β1的过度表达,则可明显降低ECM的堆积,减轻内膜肥厚的形成[8]。
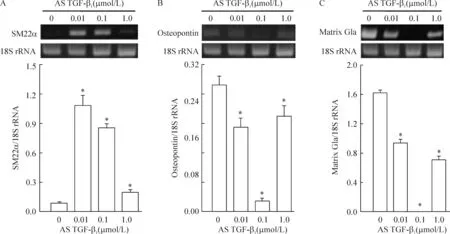
Figure 5. Effects of AS TGF-β1 on the expression of SM22α (A), osteopontin (B) and matrix Gla (C) mRNA in VSMCs after vascular injury. Mean±SD. n=4. *P<0.05 vs 0 μmol/L.
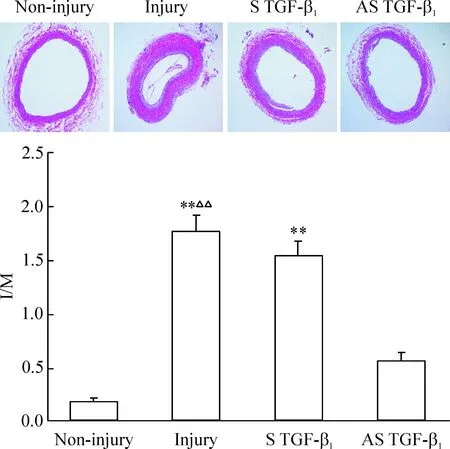
Figure 6. Effects of S and AS TGF-β1 on the intimal formation after vascular injury in SD rats (HE staining,×80). Mean±SD. n=4. **P<0.01 vs non-injury group; △△P<0.01 vs AS TGF-β1.
我们的研究结果也证实颈动脉损伤后,血管中膜并无显著变化,但血管内膜明显增厚,管腔变小。应用硫代磷酸化修饰的AS TGF-β1经皮下连续给药28 d后,显著抑制了血管损伤导致的内膜增生,管腔扩大,I/M比值下降,而S TGF-β1对血管损伤后新生内膜的增生并无明显作用。表明了AS TGF-β1由于抑制了血管损伤后VSMCs的增殖、ECM的合成和分泌,减少了ECM在血管内膜中的沉积,从而减轻了血管损伤后内膜的肥厚。以前的研究也证实了局部给予AS TGF-β1也能显著抑制血管损伤后内膜的增殖[9]。
研究发现血管损伤后,VSMCs的表型由收缩表型转变为合成表型,而合成表型的VSMCs可能合成更多的生长因子、细胞因子和血管活性物质等[10]。同时VSMCs对各种生长因子和血管活性物质,包括对TGF-β1的反应性也发生了变化,使TGF-β1由原来的抑制VSMCs增殖转而变为促进VSMCs的增殖。AS TGF-β1抑制血管损伤后VSMCs的异常增殖可能与其逆转血管损伤后VSMCs的表型转变有关。我们的研究发现应用AS TGF-β1后,VSMCs收缩表型标志物SM22α的mRNA表达水平明显增加,而代表合成表型标记物的基质Gla蛋白及骨桥蛋白的mRNA的表达水平却受到显著抑制。因此,由于AS TGF-β1使VSMCs的表型由血管损伤后的合成型恢复为未损伤时的收缩型,减少各种促生长因子、细胞因子和血管活性物质等的分泌,从而抑制了VSMCs的增殖,减少了ECM的合成和分泌,减轻了血管损伤后再狭窄的发生和发展。
总之我们所设计的AS TGF-β1能够特异性地抑制血管损伤后TGF-β1蛋白质的过度表达,逆转了VSMCs在损伤后表型的转变,从而显著抑制了血管损伤后VSMCs DNA的合成和细胞增殖,减少了ECM的合成和分泌,减轻了血管损伤后内膜的增殖。因此,AS TGF-β1有望为抑制血管损伤后新生内膜的形成,减轻PTCA术后再狭窄提供新的治疗方法和手段。
[参 考 文 献]
[1] Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis [J]. Acta Med Indones,2007,39(2):86-93.
[2] Khan R, Agrotis A, Bobik A. Understanding the role of transforming growth factor-β1in intimal thickening after vascular injury[J]. Cardiovasc Res,2007,74(2): 223-234.
[3] Yao EH, Fukuda N, Ueno T, et al. A pyle-imidazole polyamide targeting transforming growth factor-beta1 inhibits restenosis and preserves endothelialization in the injured artery[J]. Cardiovasc Res,2009,81(4):797-804.
[4] Yang Y, Mumy M, Romeo D, et al. Identification of the start sites for the 1.9- and 1.4-kb rat transforming growth factor-beta1 transcripts and their effect on translational efficiency[J]. Gene, 1998, 219(1-2): 81-89.
[5] 林志鸿,吴可贵,李庚山,等.氟伐他汀抑制球囊损伤后兔的血管内膜增殖[J].中国病理生理杂志,2005,21(1):99-103.
[6] Inouye M. Antisense RNA: its function and application in gene regulation - a review [J]. Gene, 1988, 72(1-2): 25-34.
[7] Bobik A. Transforming growth factor-βs and vascular disorders [J].Arterioscler Thromb Vasc Biol,2006,26(8):1712-1720.
[8] Merrilees M, Beaumout B, Scott L, et al. Effect of TGF-β1antisenseS-oligonucleotide on synthesis and accumulation of matrix proteoglycans in balloon catheter-injured of rabbit carotid arteries[J]. J Vasc Res, 2000, 37(1):50-60.
[9] Sun DX, Liu Z, Tan XD, et al. Nanoparticle-mediated local delivery of an antisense TGF-β1 construct inhibits intimal hyperplasia in autogenous vein grafts in rats[J]. PLoS One,2012, 7(7): e41857.
[10] Chaabane C, Otsuka F, Virmani R, et al. Biological responses in stented arteries[J]. Cardiovasc Res, 2013, 99(2):353-363.
猜你喜欢
昆明医科大学学报(2021年12期)2021-12-30
小学生学习指导·低年级(2021年6期)2021-09-10
考试与评价·七年级版(2021年4期)2021-08-14
现代仪器与医疗(2021年2期)2021-07-21
天津医科大学学报(2021年1期)2021-01-26
心肺血管病杂志(2020年5期)2021-01-14
小学阅读指南·低年级版(2018年5期)2018-11-02
中华骨与关节外科杂志(2017年1期)2017-05-17
中国男科学杂志(2016年5期)2016-12-01
中国康复理论与实践(2015年10期)2015-12-24
