
高效液相色谱法测定葛根芩连片中的葛根素
2014-07-24 18:57:09王海鸣严志红
化学分析计量 2014年4期
王海鸣,严志红
(1.广州广电计量检测股份有限公司,广州 510656; 2.广东药学院,广州 510006)
高效液相色谱法测定葛根芩连片中的葛根素
王海鸣1,严志红2
(1.广州广电计量检测股份有限公司,广州 510656; 2.广东药学院,广州 510006)
建立高效液相色谱测定葛根芩连片中葛根素含量的方法。以体积分数50%的甲醇为提取液对样品超声提取20 min,采用DiamonsilTMC18(250 mm×4.6 mm ,5μm)色谱柱,以甲醇-乙腈-水(体积比8∶12∶80)为流动相,流速为1.0 mL/min,检测波长为250 nm,柱温为30.0℃,进样量体积10 μL。在最佳实验条件下,葛根素与其它物质能完全分离,葛根素的质量浓度在5.43~543.2 μg/mL范围内与色谱峰面积呈良好的线性,线性相关系数r=0.999 9,方法检出限为3.50 μg/mL (S/N=3)。方法加标回收率为100.0%,测定结果的相对标准偏差为1.6% (n=6)。该方法简单、快速、重现性好,适用于葛根芩连片中葛根素的测定。
高效液相色谱;葛根芩连片;葛根素
葛根芩连片是由葛根、黄芩、黄连组成的中药制剂,具有清热解毒、止泻止痢的作用,临床用于治疗泄泻、痢疾、身热烦渴等症[1],其中葛根为方中君药,而葛根素为葛根的主要活性成分之一,具有活血通脉,解肌退热,升阳止泻的功能[2]。相关制剂在2010年版《中国药典》中收载,葛根素为葛根质量控制的主要成分,对其提取及含量测定方法进行研究具有重要意义。
葛根芩连片中葛根素,常以加热回流进行提取后制成供试品溶液由高效液相色谱法进行测定[3-5],但提取方法较繁琐[6]。笔者采用超声提取-高效液相色谱法进行了葛根芩连片中葛根素的测定,优选了提取溶剂和提取时间。该方法简便、提取完全,定量分析结果的精密度、准确度较高。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:由高压泵(LC-10ATvp型)、自动进样器(SIL-20A型)、柱温箱(CTO-10Asvp型)、二极管阵列检测器(SPD-M10Avp型)、工作站(LCsolution型)组成,日本岛津公司;
超声波清洗仪:DL-360A型,日本岛津公司;
葛根素对照品:编号为110752-200912,中国药品生物制品检定研究院;
葛根芩连片:商品名为畅康宁,批号分别为090301,090501,090701,山西省恒山中药有限责任公司;
甲醇、乙腈:色谱纯,德国默克公司;
色谱使用双蒸水,其它实验用水为蒸馏水;
实验所用其它试剂均为分析纯。
1.2 色谱条件
色谱柱:DiamonsilTMC18(250 mm×4.6 mm,5 μm),流动相:甲醇-乙腈-水(体积比8∶12∶80),流速为1.0 mL/min;检测波长:250 nm;柱温:30℃;进样体积:10 μL。
1.3 溶液制备
(1)葛根素对照品溶液的制备。精密称取葛根素对照品适量,以50%甲醇溶解并定容于50 mL的容量瓶中,质量浓度为543.2 μg/mL,使用时按需稀释;
(2)葛根素供试品溶液的制备。取葛根芩连片20片,研细得粉末,精密称取粉末0.15 g,置于50 mL的容量瓶中,加入50%甲醇25 mL,振摇溶解,超声20 min,放置冷却,以50%甲醇定容,摇匀,滤过,取续滤液,得葛根素供试品溶液。
(3)葛根素阴性对照溶液的制备。根据葛根芩连片药品说明书及厂家提供的制剂配方,配制不含葛根的阴性对照粉末。精密称取阴性对照0.15 g,按照上述(2)方法制得葛根素阴性对照溶液。
2 结果与讨论
2.1 检测波长
根据葛根素标准溶液的二极管阵列紫外图谱可知,葛根素在250 nm处有最大吸收,因此实验选择250 nm为葛根素的检测波长。
2.2 提取溶剂
设置超声提取时间为20 min,其它条件保持不变,分别试验了以体积分数10%,30%,50%,70%,100%甲醇为提取剂时超声提取效果(以葛根素峰面积为指标)。结果表明,经上述不同体积分数甲醇提取后,葛根素平均色谱峰面积分别为2.25×107,2.36×107,2.38×107,2.26×107,1.94×107,因此超声提取溶剂选择50%甲醇。
2.3 提取时间
以50%甲醇为提取溶剂,其它条件保持不变,分别考察超声提取时间为10,20,30,40,50 min时的提取效果(以葛根素峰面积为指标)。结果显示,在上述不同提取时间下,葛根素平均色谱峰面积为2.14×107,2.44×107,2.46×107,2.46×107,2.45×107。考虑到20 min以后葛根素峰面积变化不大,实验选择超声提取时间为20 min。
2.4 流动相
流动相为甲醇-水,峰形较差,出峰比较快,容易受到其它杂质的干扰;当选择甲醇-乙腈-水(8∶12∶80)为流动相,流速为1.0 mL/min时,色谱峰形好,葛根素的分离度符合要求,灵敏度高,保留时间适中。
2.5 专属性试验
分别取葛根素对照品溶液、葛根素供试品溶液和葛根素阴性对照溶液进样分析,所得色谱图见图1~图3。由图1~图3可见,在选定的色谱条件下,供试品中葛根素与其它杂质能完全分离且阴性对照无干扰,葛根素保留时间约为15.4 min,理论塔板数以葛根素计不低于3 000。
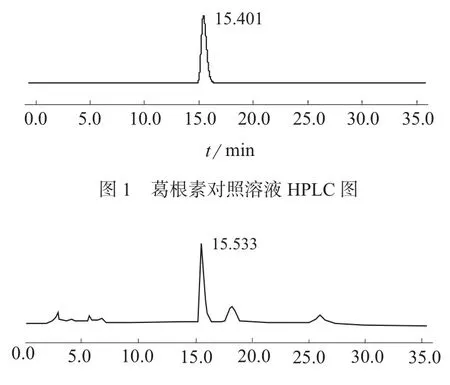
图2 葛根芩连片供试溶液HPLC图
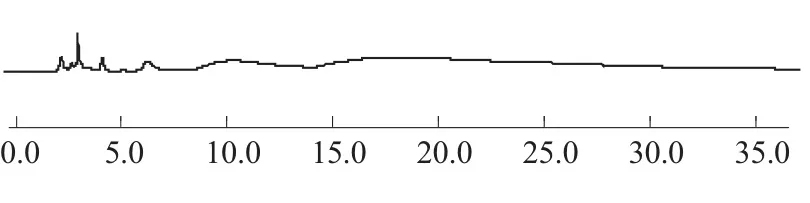
图3 不含葛根的阴性对照溶液HPLC图
2.6 标准曲线方程及检出限
分别精密移取葛根素对照品溶液适量,适当稀释得质量浓度为543.2,271.6,135.8,54.32,27.16,13.58,5.432 μg/mL的系列标准溶液,在1.2色谱条件下分别进样分析。以葛根素色谱峰面积(A)为纵坐标,葛根素质量浓度(c)为横坐标绘制标准曲线,得回归方程为A=38 248c-44 170,相关系数r=0.999 9,线性范围为5.43~543.2 μg/mL。以3倍标准偏差计算(S/N=3)检出限为3.50 μg/mL。
2.7 供试品溶液稳定性试验
对批号090501样品按1.3(2)制备供试品溶液一份,在1.2色谱条件下,分别按时间间隔0,2,4,6,8,10,12 h进样分析,结果见表1。由表1可知,测定结果的相对标准偏差为1.8%,表明供试品溶液在12 h内是稳定的。

表1 供试品溶液稳定试验结果
2.8 精密度试验
精密称取批号090501的葛根芩连片样品共6份,按照1.3(2)方法制备供试品溶液,在1.2色谱条件进行测定,结果见表2。由表2可知,葛根素含量测定结果的相对标准偏差为1.5%,说明所建方法精密度良好。

表2 精密度试验结果(n=6)
2.9 回收试验
精密称取葛根芩连片样品(批号090501)6份,按1.3(2)方法制备供试品溶液,进样分析。然后分别加入质量浓度为543.2 μg/mL的葛根素对照品溶液2.00 mL,进行加标回收试验,结果见表3。由表3可知,平均回收率为100.0%,说明方法的准确度较高。
2.10 样品测定
按1.3(2)方法将3批葛根芩连片样品(090301,090501,090701)分别制成供试品溶液,每批3份,分别进样测定,计算样品中葛根素含量,结果见表4。由表4可知,3批样品中的葛根素含量相差不大,测定结果的相对标准偏差均小于2.0%。由此可见方法测量的精密度良好。

表3 回收试验结果(n=6)

表4 样品中葛根素的含量测定结果(n=3)
3 结语
采用超声提取-高效液相色谱法测定葛根芩连片中有效成分葛根素含量,优选了提取溶剂和提取时间,该法具有快速简便、稳定性好、结果精确高的特点,可作为葛根制剂质量控制的方法。
[1]唐波,王小宁.葛根芩连汤精制颗粒治疗湿热泄泻临床研究[J].时珍国医国药,2010,10(11): 920-922.
[2]宋炜,高卫华.葛根素的药理作用解析及临床用途[J].中外医疗,2011(4): 139.
[3]吴永芹,冯凯,周慧,等.HPLC法同时测定葛根芩连片中葛根素和黄芩苷的含量[J].中国药事,2012,26(3): 282-284.
[4]张隽,张祥伟,黄添友,等.葛根芩连汤超微饮片与传统饮片葛根素溶出量及药效作用对比研究[J].时珍国医国药,2011,22(2): 358-361.
[5]邓开英,王晓琳,夏永鹏,等.葛根芩连片中3种活性成分的HPLC法测定[J].中国医药工业杂志,2011,42(10): 784-786.
[6]骆红飞,黄丽娜.反相高效液相色谱法测定葛根芩连丸中葛根素的含量[J].浙江中医杂志,2013,48(3): 225-226.
Determination of Puerarin in Gegenqinlian Tablet by HPLC
Wang Haiming1,Yan Zhihong2
(1. Guangzhou GRG Metrology & Test Co. Ltd., Guangzhou 510656,China;2.Guangdong Pharmaceutical University,GuangZhou 510006,China)
HPLC method was established for the determination of puerarin in Gegenqinlian tablet. The sample was extracted by ultrasonic with 50% methanol and extraction time of 20 min. The diamonsilTMC18(250 mm ×4.6 mm 5μm) column was used with methanol-acetonitrile-water (volue rate was 8∶12∶80) as the mobile phase,the flow rate was 1.0 mL/min,detection wavelength was 250 nm,column temperature was 30.0℃ and injection volume was 10 μL. HPLC method could completely separated puerarin from other substances under the optimum experimental conditions. The concentration of puerarin was linear with peak area in the range of 5.43-543.2μg/mL,the linear correlation coefficient was 0.999 9,and the detection limit was 3.50 μg/mL. The average recovery of method was 100.0% and the relative standard deviation was 1.6% (n=6). The method is simple,rapid,reproducible,and it is suitable for the determination of puerarin in Gegenqinlian tablet.
HPLC; Gegenqinlian tablet; puerarin
O657.7
A
1008-6145(2014)04-0070-03
10.3969/j.issn.1008-6145.2014.04.021
联系人:严志红;E-mail: yzhxsp@aliyun.com
2014-05-08
猜你喜欢
Cancer Biology & Medicine(2024年1期)2024-02-24 09:56:46
昆明医科大学学报(2021年4期)2021-07-23 01:21:32
智富时代(2019年2期)2019-04-18 07:44:42
中成药(2018年6期)2018-07-11 03:01:04
中成药(2018年5期)2018-06-06 03:11:53
石油地球物理勘探(2017年4期)2017-12-18 07:14:39
中成药(2017年6期)2017-06-13 07:30:35
中国老区建设(2016年1期)2016-02-28 09:31:56
中国中医药现代远程教育(2016年20期)2016-02-14 22:21:47
学习月刊(2015年15期)2015-07-09 05:38:34
