
石墨炉原子吸收分光光度法测定固体废物中铍和钼
2014-07-24 18:57任兰杜青陆喜红
化学分析计量 2014年4期
任兰,杜青,陆喜红
(南京市环境监测中心站,南京 210013)
石墨炉原子吸收分光光度法测定固体废物中铍和钼
任兰,杜青,陆喜红
(南京市环境监测中心站,南京 210013)
建立了石墨炉原子吸收法测定固体废物中铍和钼的方法。采用盐酸-硝酸-氢氟酸-高氯酸消解样品,钯盐作基体改进剂,消除了基体干扰。铍、钼的质量浓度分别在0~4.0,0~50.0 μg/L范围内与吸光度呈良好的线性,线性相关系数均为0.999 6,检出限分别为0.03,0.2 μg/g。实际样品加标回收率为82.5%~117.0%,测定结果的相对标准偏差为6.6%~10.4% (n=6)。该方法选择性强、灵敏度高,测定结果准确,满足固体废物全量分析要求。
石墨炉原子吸收法;固体废物;铍;钼
随着经济发展和城市化水平的提高,固体废物的产量迅速增长,且种类繁多,已成为人类社会和经济健康发展的一大难题[1],含有金属元素的固体废物给环境带来的污染也越来越重。目前我国固体废物控制标准主要有《危险废物鉴别标准浸出毒性鉴别》[2]和《危险废物鉴别标准毒性物质含量鉴别》[3]。关于固体废物中金属元素的测定,在国标方法中只规定了铜、锌、铅、镉、铬、镍等几种金属元素的测定方法,并且测定对象为固体废物浸出液。固体废物中毒性物质含量测定目前尚无国标方法。为了更好地适应社会、经济、科学技术的发展,有必要对固体废物中金属元素全量的测定方法进行研究。
环境样品中铍和钼的测定方法有分光光度法、原子吸收分光光度法、ICP-AES、ICP-MS等,石墨炉原子吸收法是痕量分析的经典方法,分析成本较ICP-MS低,是单元素痕量分析的首选。ICP-AES,ICP-MS是当前较先进的技术,对于多元素痕量分析具有优势,但仪器价格昂贵,分析成本较高。固体废物中铍和钼的测定鲜有报道。笔者采用盐酸-硝酸-氢氟酸-高氯酸对固体废物进行消解,以钯盐作基体改进剂,用石墨炉原子吸收分光光度法测定固体废物中全量铍、钼。该方法灵敏度高、测定结果准确可靠。
1 实验部分
1.1 主要仪器与试剂
石墨炉原子吸收光谱仪:AA240Z型,塞曼扣背景,美国Varian公司;
铍空心阴极灯、钼空心阴极灯:美国Varian公司;
电热板:EG20B型,美国LabTech公司;氩气:含量不低于99.99%;
铍标准储备液:1 000 μg/mL(扩展不确定度4 μg/mL,介质10% HNO3),钢铁研究总院,国家钢铁材料测试中心;
钼标准储备液:1 000 μg/mL(扩展不确定度为4 μg/mL,介质为5% H2SO4),钢铁研究总院,国家钢铁材料测试中心;
土壤标准参考样:GSS-5(GBW-07405),地矿部物化探所测试所;
盐酸、硝酸、氢氟酸、高氯酸:优级纯;氯化钯:分析纯;硝酸镁:优级纯;
氯化钯溶液:称取0.34 g氯化钯,加少量水和1 mL硝酸,于50℃加热溶解,用水定容至100 mL;
氯化钯-硝酸镁混合溶液:称取0.25 g氯化钯,加少量水和1 mL硝酸,于50℃加热溶解,称取0.3 g硝酸镁溶于少量水中,将两种溶液混合,用水稀释定容至100 mL;
实验用水为二次去离子水。
1.2 仪器工作条件
石墨炉原子吸收光谱仪工作参数见表1。

表1 仪器工作条件
1.3 样品前处理
将固体废物样品经自然风干并碾磨后,过150 μm(100目)筛,制得粉末试样。称取0.3 g(精确至0.1 mg)粉末试样于50 mL聚四氟乙烯坩埚中,用水润湿后加入l0 mL盐酸,于通风橱内的电热板上低温加热,使样品初步分解,待蒸发至约剩3 mL时,加入5 mL硝酸,5 mL氢氟酸,加盖后于电热板上120~130℃加热0.5~1 h,开盖加入2 mL高氯酸,再加盖150~160℃加热1 h左右,开盖,驱赶白烟并蒸至内容物呈不流动状态(趁热观察)。视消解情况,可再补加3 mL硝酸、3 mL氢氟酸、1 mL高氯酸,重复以上消解过程。取下坩埚稍冷,加入1 mL硝酸溶液(1+1),温热溶解可溶性残渣,转移至50 mL容量瓶中,冷却后用水定容至标线,摇匀。
2 结果与讨论
2.1 石墨管的选择
普通石墨管耐高温性能差,寿命短,灵敏度低。钼在高温下易形成碳化物,记忆效应显著[4],实验使用热解涂层石墨管大大减少碳化物的形成,提高了灵敏度。
2.2 石墨炉升温程序的优化
灰化和原子化阶段尤为重要。通过试验绘制灰化曲线和原子化曲线,以铍为例图1~图4为灰化温度、时间及原子化温度、时间曲线。最终得到仪器最佳升温程序,见表2。

图1 铍灰化温度曲线

图2 铍灰化时间曲线

图3 铍原子化温度曲线

图4 铍原子化时间曲线

表2 石墨炉升温程序
2.3 消解体系的选择
固体废物全量消解目前没有标准方法,国内文献对固体废物的消解有硝酸-氢氟酸-高氯酸混酸体系[5]、硝酸-氢氟酸-过氧化氢混酸体系[6]。考虑到固体废物样品来源的多样性,有的有机物含量高(污水处理厂污泥),有的有硅晶结构(如矿业固体废物),实验采用盐酸-硝酸-氢氟酸-高氯酸混酸体系消解固体废物。
2.4 共存元素干扰
相对误差在±10%之内时,考察共存离子对测定的干扰,结果表明5 000倍的Cu,Zn,Ni,Pb,Cd,Cr,Mn ,Ba,25 000倍的Ti,50 000倍的K,Na,Ca,Al不干扰2.0 μg/L铍的测定;1 000倍的Ag,Al,B,Ba,Be,Bi,Cd,Co,Fe,Hg,Cr,Cs,Cu,Mg,Mn,Ni,Pb,Sb,Se,Sn,Sr,Ti,Tl,V,Zn和10 000倍K,Na,Ca,Mg不干扰10.0 μg/L钼的测定。
2.5 基体改进剂的选择
基体改进剂的加入可以消除干扰、提高灰化温度和分析灵敏度,国内文献报道石墨炉分析基体改进剂有硝酸镁[4,7]、硝酸锂[8]、硝酸铝、硝酸钯、磷酸二氢铵、抗坏血酸等[4],美国EPA200.9[9]推荐以钯盐加硝酸镁作基体改进剂。对硝酸镁、硝酸铝、氯化钯、磷酸二氢铵-硝酸镁、氯化钯-硝酸镁、氯化钯-抗坏血酸基体改进剂(进样量为5 μL)进行了试验,分别对实际固废样品中的铍、钼进行测定。测定结果表明,以氯化钯作为测定铍的基体改进剂,以氯化钯-硝酸镁作为测定钼的基体改进剂效果最好。
2.6 标准曲线方程
用1%硝酸将铍标准储备溶液逐级稀释配制成4.0 μg/L铍工作溶液,仪器自动配制成0.0,1.0,2.0,3.0,4.0 μg/L的系列标准溶液,分别进行测定,以吸光值y对铍的质量浓度x (μg/L)绘制标准曲线,得回归方程为y=0.114x+0.015 2,相关系数r=0.999 6,线性范围为0~4.0 μg/L。
用1%硝酸将钼标准储备溶液逐级稀释配制成50.0 μg/L钼工作溶液,仪器自动配制0.0,5.0,10.0,20.0,30.0,50.0 μg/L的系列标准溶液,以吸光度y对钼的质量浓度x(μg/L)绘制标准曲线,得回归方程为y=0.010 3x+0.001 6,相关系数r=0.999 6,线性范围为0~50.0 μg/L。
2.7 方法检出限
根据HJ 168-2010规定[10],采用低浓度加标方式计算方法检出限。分别取7份0.5 μg/L铍标准溶液和7份1.5 μg/L钼标准溶液,按样品分析全过程测定,以MDL=t(n-1,0.99)s(s为测定结果的标准偏差)计算得铍、钼的检出限分别为0.03,0.2 μg/g(以称样0.300 0 g、定容体积50 mL计)。
2.8 准确度试验
称取6份土壤标准参考样GSS-5,按1.3方法消解、测定,结果见表3。由表3可见,铍和钼含量的测定值均在保证值误差范围内,由此可见该方法准确度较高。

表3 准确度试验结果
2.9 精密度试验
采集6种不同固体废物样品(1#~6#),按1.3方法处理后进行测定,结果见表4。由表4可知,实际固废样品测定结果的相对标准偏差为6.60%~10.4%,表明方法的精密度良好。
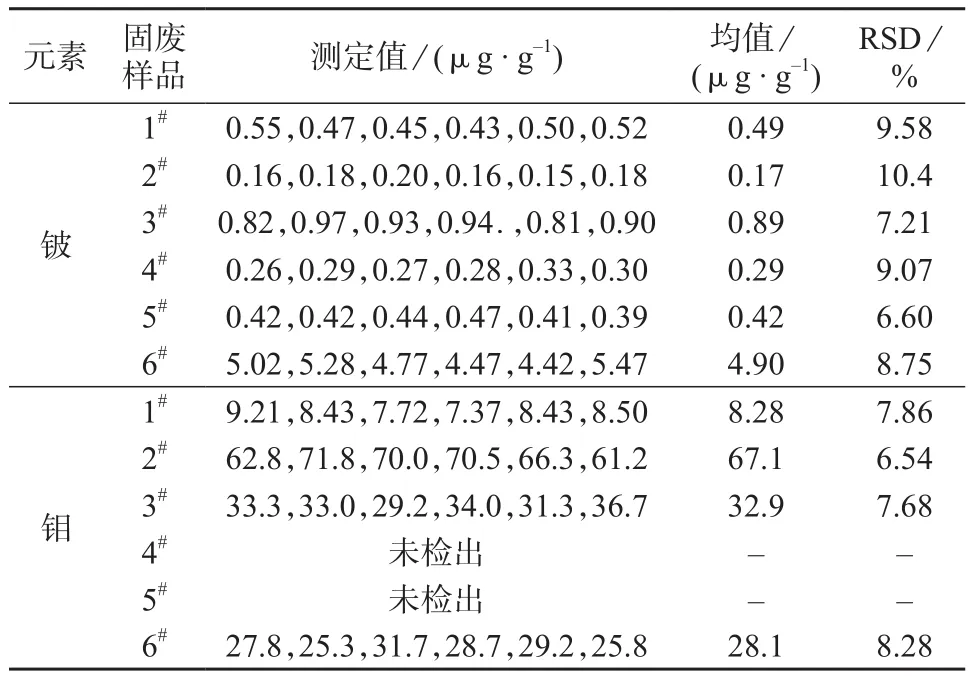
表4 精密度试验结果
2.10 回收试验
准确称取实际固体废物样品0.300 0 g,按实验方法测定后,进行铍、钼加标回收试验,结果见表5。由表5可知,铍、钼加标回收率为82.5%~117.0%,满足日常分析的质量控制要求。

表5 加标回收试验结果
3 结语
固体废物中铍、钼的全量测定目前尚无国家标准方法,采用石墨炉原子吸收法测定固废样品的铍、钼,以盐酸-硝酸-氢氟酸-高氯酸混酸体系消解样品,以氯化钯、氯化钯-硝酸镁为基体改进剂,方法灵敏度高、测定结果准确可靠,能满足固体废物全量分析要求,适用性强,易于推广应用。
[1]胡涛.我国固体废物的管理体制问题分析[J].环境科学研究,2006,19(增刊): 33.
[2]GB 5085.3-2007 危险废物鉴别标准浸出毒性鉴别[S].
[3]GB 5085.6-2007 危险废物鉴别标准毒性物质含量鉴别[S].
[4]邓勃.原子吸收光谱分析[M].北京:化学工业出版社,2004.
[5]时玉珍.同一固废全消解液中Cu、Pb、Zn、Cd、Mn的原子吸收光谱测定方法研究[J].水泥技术,2011(1): 102.
[6]陈波,陈素兰.微波消解/ICP-AES法分析冶炼厂固废中重金属[J].环境保护科学,39(4): 121.
[7]宋建刚.硝酸镁对GFAA 法测定微量铍的基体改进效应[J].光谱实验室,2006,23 (6): 1 299-1 301.
[8]林琳,梁东松,韩华云,等.涂钼石墨管-石墨炉源自吸收法测定人发中的微量铍[J].分析科学学报,2005,21(3): 353-354.
[9]EPA METHOD 200.9 Trace elements in water,solids,and biosolids by stabilized temperature graphite furnace atomic absorption spectrometry[S].
[10]HJ 168-2010 环境监测分析方法标准制修订技术导则[S].
Determination of Beryllium and Molybdenum in Solid Wastes by Graphite Furnace Atomic Absorption Spectrometry
Ren Lan, Du Qing, Lu Xihong
(Nanjing Environmental Monitoring Center, Nanjing 210013, China)
A method for determination of beryllium and molybdenum in solid wastes by graphite furnace absorption spectrometry was established. Hydrochloric acid, nitric acid, hydrofluoric acid and perchloric acid were selected to digest samples, and palladium salt was choosed as matrix modifier to eliminate the matrix interference. The concentration of beryllium and molybdenum showed a linear dependence on absorbence in the range of 0-4.0 μg/L, 0-50 μg/L, respectively, with the correlation coefficient of 0.999 6. The detection limits of beryllium and molybdenum were 0.03, 0.2 µg/g, respectively. The relative standard deviation of detection results was 6.6%-10.4% (n=6), and the recovery was between 82.5% and 117.0%. Therefore, the method is sensitive, which accuracy can meet the requirement of the full analysis of solid waste.
graphite furnace atomic absorption spectrometry; solid waste; beryllium; molybdenum
O657.3
A
1008-6145(2014)04-0038-04
10.3969/j.issn.1008-6145.2014.04.011
联系人:任兰;E-mail: renlan001@126.com
2014-05-09
猜你喜欢
中学生数理化·高一版(2020年11期)2020-12-14
中学化学(2019年2期)2019-07-08
发明与创新·中学生(2019年6期)2019-06-26
浙江化工(2018年1期)2018-02-03
河北地质(2016年2期)2016-03-20
化工设计通讯(2016年5期)2016-03-13
中国资源综合利用(2016年6期)2016-01-22
食品研究与开发(2015年20期)2015-07-22
无机化学学报(2014年6期)2014-02-28
舰船电子工程(2012年8期)2012-07-11
