
聚吡咯改性电沉积Sn负极材料的电化学性能
2014-07-14 05:19:38温兆银吴梅芬杨建华
无机化学学报 2014年6期
彭 鹏 温兆银 刘 宇 吴梅芬 杨建华
(中国科学院上海硅酸盐研究所能量转换重点实验室,上海 200050)
0 引 言
锂离子电池因具有能量密度高、重量轻、环境友好等优点已广泛应用于电子器件市场。Sn因能与锂形成Li22Sn5储锂产物,具有比商用石墨电极(372 mAh·g-1)高得多的比容量(987 mAh·g-1)[1,2]而引起了广泛的关注。该类材料还具有脱嵌锂电压低(ca.0.8 V vs Li/Li+)、与电解液的反应活性低等优点,有利于发展成为大功率安全性电源之一。Sony公司推出的Sn-Co-C锂离子电池进一步推动了锡基合金负极材料的研究,但这类材料在电化学脱嵌锂过程中存在着较大的体积效应,产生的机械应力会使电极活性物质破碎,丧失与集流体的电接触,从而造成电极电化学循环性能的衰减[3]。目前解决该问题的方法有很多,包括引入其他活性或者惰性相形成合金体系或者其他导电材料形成复合体系等。Tabuch等[4]通过共电镀的方式得到Sn-Sb-Co三相合金,具有580 mAh·g-1的可逆比容量,其他合金体系还有Sn-Cu[5]、Sn-Sb[6,7]、Sn-Ag[8]、Sn-Ni[9]、Sn-Ca[10]等。Wang 等[11]将Sn分散在有序介孔石墨碳层中,利用碳层骨架形成的有序导电网络缓解充放电过程中的体积效应,100次循环后剩余560 mAh·g-1稳定比容量。本工作在铜箔上直接电沉积Sn负极,避开常规操作中粘结剂的使用,简化工艺程序,同时利用导电聚合物聚吡咯(PPy)对沉积后的电极片进行表面修饰改性,聚吡咯具有良好的弹性和粘附性,可形成稳定牢固的导电通道[12-13],能有效缓解合金负极的体积效应同时增加材料的导电性,保持活性材料与集流体的电接触,改善Sn负极的电化学性能。
1 实验部分
1.1 电极材料的制备
1.1.1 Sn 负极的电沉积
以厚度15 μm铜箔为工作电极,厚度150 μm镍片为对电极,在自制简易电解槽(如图1)中恒电流密度(0.35 mA·cm-2)恒温(50 ℃)沉积 2h,电解液为SnCl2、NaF(各 30 g·L-1,盐酸调节 pH=4~5),取出电极,经去离子水洗涤、干燥,简记为Sn。
不同浓度 NiCl2(0、10、25、50、100 g·L-1) 作为添加剂分别溶解于SnCl2-NaF电解液中,重复相同的步骤,沉积10 min,电极经去离子水洗涤、干燥,分别简记为 Sn(Ni)-0、Sn(Ni)-10、Sn(Ni)-25、Sn(Ni)-50、Sn(Ni)-100。
1.1.2 聚吡咯修饰Sn的电极制备
经去离子水洗涤的电极悬挂浸入预先超声分散均匀的吡咯悬浮液中,0℃逐滴缓慢加入氧化剂(NH4)2S2O8(n吡咯:n(NH4)2S2O8=1∶1),滴加完毕后静置 3 h,取出电极,经去离子水洗涤、干燥。

图1 自制简易电解槽示意图Fig.1 Schematic diagram of the electrolysis setup
1.2 电极材料的表征及电化学性能测试
材料的结构由X射线衍射(Riguka,D/max2550VB3+/PC 型,Cu 靶,Kα1射线,λ=0.154 056 nm,扫描电压 40 kV,电流 100 mA,扫描范围 10°~80°,扫描速度为 6°·min-1)表征,JSM-6700 型扫描电子显微镜分析电极片的表面形貌;元素分布由INCA型能谱仪分析(英国牛津)。将沉积的电极片直接切成圆片(φ 14 mm),以金属锂为对电极和参比电极,Celgard2325 为隔膜,1 mol·L-1LiPF6的碳酸乙烯酯 (ethylene carbonate,EC):碳酸二甲酯(dimethyl carbonate,DMC)(物质的量的比为 1∶1)有机溶液(其中含有2wt%碳酸亚乙烯酯)为电解质,组装成2025扣式电池。LAND CT2001A充放电仪恒流模式进行充放电,电流密度为120 mA·g-1,电压范围为0.1~1.5 V。Autolab PGSTAT302型电位-恒电流电化学工作站分析电化学循环伏安曲线和电化学交流阻抗谱,循环伏安的扫描电压范围为 0.05~1.5 V,扫描速度为0.5 mV·s-1,交流阻抗谱测试频率范围为10-2~105Hz,交流振幅为5 mV。
2 结果与讨论
2.1 电解液成分的影响
图2显示的是在电解液中添加了不同浓度NiCl2后沉积电极的XRD图,除去铜箔的特征衍射峰 ,Sn(Ni)-10、Sn(Ni)-25、Sn(Ni)-50、Sn(Ni)-100 都 在30.3°出现了对应于四方结构 Sn(a=b=0.538 1 nm,c=0.318 72 nm,PDF No.04-0673)的(200)特征衍射峰。所有沉积电极的XRD图中均没有杂相衍射峰,说明电极表面是纯相Sn。而没有添加NiCl2的沉积电极Sn(Ni)-0的XRD图中没有Sn的特征衍射峰出现,说明在短时间内电极表面没有Sn颗粒析出。添加了NiCl2有利于缩短Sn负极的沉积时间,提高了沉积时率。

图2 不同浓度NiCl2沉积10 min的电极XRD图Fig.2 XRD patterns of anodes deposited for 10 min with different concentrations of NiCl2
在电解液中添加不同浓度NiCl2后沉积10 min的电极形貌如图3所示。比较发现,电解液中没有添加NiCl2沉积的电极表面没有Sn颗粒析出,而添加了NiCl2的4种电解液中,沉积电极的表面均析出了一层细小的纳米Sn颗粒同时伴随着类似Sn空心管颗粒。随着NiCl2浓度增加,电极表面析出的Sn颗粒越多颗粒粒径随之长大,空心管也逐渐变大。这是因为NiCl2浓度越大,析出Sn颗粒速率越快,沉积在铜箔表面的Sn颗粒生长也越快,因而粒径越大。但是当NiCl2浓度过高,例如Sn(Ni)-100的表面形貌,不仅表面沉积的纳米Sn颗粒粒径分布不均,甚至Sn空心管开始消失,而一端开口的球形颗粒开始增多,这是因为NiCl2浓度过高导致Sn颗粒在电极表面析出速率过快,颗粒来不及自组装成空心管,更容易团聚成球,这对利用Sn空心管状结构来缓解体积效应是不利的。

图3 不同浓度NiCl2沉积10 min的电极表面SEM图Fig.3 SEM images of anodes deposited for 10 min with different concentrations of NiCl2
图4中对应的EDS图也显示添加了不同浓度NiCl2沉积的电极表面EDS相似,都存在Sn元素的峰,而没有添加NiCl2沉积的电极Sn(Ni)-0表面没有Sn元素峰存在。此外EDS图中除了铜箔对应的Cu元素以及导电胶的C元素外,没有Ni元素峰出现,说明电极表面颗粒是纯相Sn。
值得注意的是EDS谱中都没有干扰元素F出现,说明NaF不参与电沉积反应。SnCl2溶于水易分解,NaF可与SnCl2形成配合物[SnClF2]增加溶解度,同时能降低Sn2+的析出电位。此外,在镍片为对电极的沉积体系中,电解液中添加NiCl2有助于抑制镍对电极发生不必要的副反应进入电解液中,从而影响沉积效果。
2.2 聚吡咯表面修饰
结合沉积时效和沉积电极表面形貌,选择Sn(Ni)-0、Sn(Ni)-50沉积 2 h,继续研究 NiCl2对沉积电极电化学性能的影响,对应电极分别简记为Sn-2h、Sn(Ni)-2h,同时分别对沉积电极的表面进行聚吡咯修饰,简记为 Sn-2h-PPy、Sn(Ni)-2h-PPy。
2.2.1 沉积电极的表面形貌表征
图5为沉积电极和聚吡咯修饰后电极表面的SEM图,图5(a)中Sn-2h析出的细小纳米颗粒均匀分布在铜箔基体表面,图5(b)中Sn(Ni)-2h表面呈现空心管周围伴随着微米级球状颗粒的形貌,空心管内径约 2.5 μm,外径约 4.0 μm,长度在 20 μm 以上。聚吡咯修饰后,Sn-2h-PPy电极表面覆盖了一层粒径分布集中且分散性好的球状聚吡咯纳米颗粒(粒径小于500 nm),Sn(Ni)-2h-PPy电极表面的空心管也覆盖着球状聚吡咯纳米颗粒。
2.2.2 电化学性能研究
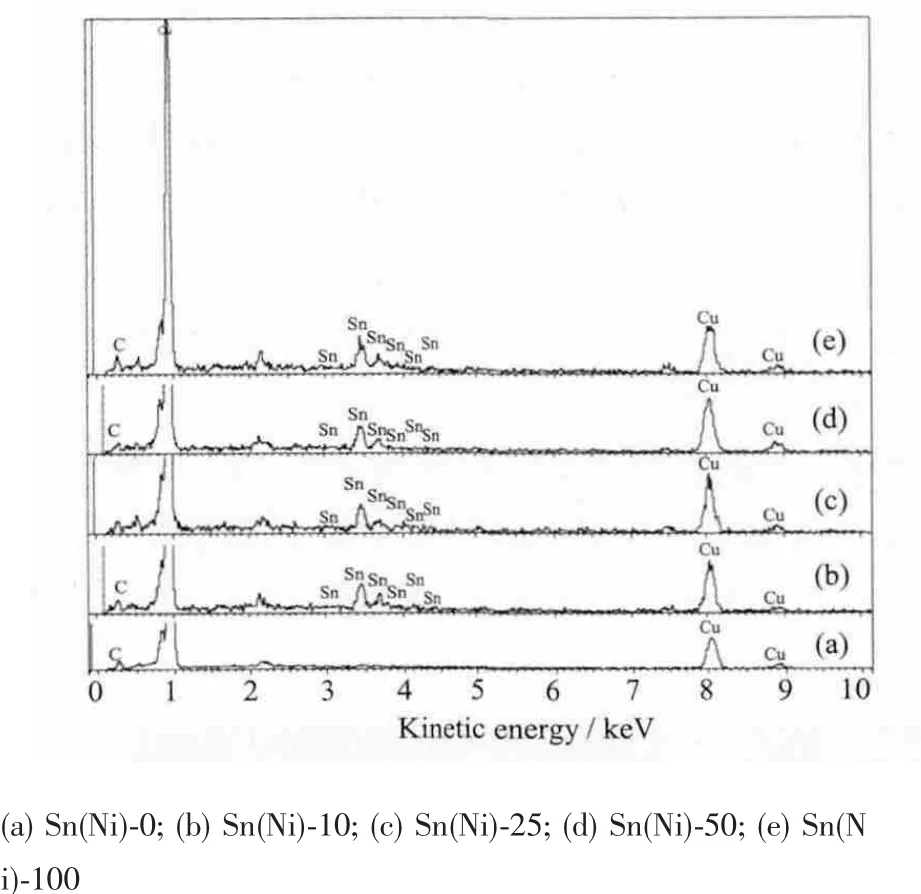
图4 不同浓度NiCl2沉积10 min的电极表面EDS图Fig.4 EDS of anodes deposited for 10 min with different concentrations of NiCl2
图6(a)循环寿命曲线显示Sn-2h在首次循环后可逆比容量迅速衰减殆尽,且首次放电比容量也较低,这是由于纳米级Sn颗粒单独分散在铜箔基体表面,相互间的作用力小,周围没有其他相能够提供保护效用,缓解循环过程中合金化/去合金化产生的巨大体积效应,从而造成Sn颗粒发生破碎,从基体上脱落,丧失与集流体的电接触,使得容量迅速衰减。Sn(Ni)-2h电极由于微米级球状颗粒的大量存在,这些颗粒的体积效应不能被自身的结构所消化,容易破碎失去脱嵌锂活性,但是另外由于Sn空心管结构的存在,这种结构对体积效应存在一定程度的缓解作用,即使在没有其他相的辅助保护下,仍然能保有一些电化学活性,故Sn(Ni)-2h电极在60次循环后仍剩余一部分可逆比容量,达184.3 mAh·g-1。
利用PPy表面修饰后Sn-2h-PPy、Sn(Ni)-2h-PPy电极的电化学循环稳定性能都有了大幅度的提升。在60次循环后分别剩余397.5、440.6 mAh·g-1可逆比容量。
从图6(a)的内插图可看出虽然Sn-2h-PPy的可逆比容量在循环初期高于Sn(Ni)-2h-PPy,但随着循环次数的增加可逆比容量呈现小幅度缓慢衰减的变化趋势,在57次循环后可逆比容量已经低于Sn(Ni)-2h-PPy了且仍没达到稳定平衡态,而Sn(Ni)-2h-PPy从30次循环以后可逆比容量基本保持稳定,因此Sn(Ni)-2h-PPy电极在保持高可逆比容量的同时循环稳定性也非常优越,说明表面修饰了聚吡咯对电极的电化学循环性的确有一定的改善,能够提高循环的稳定性。

图5 沉积电极和修饰电极表面的SEM图Fig.5 SEM images of deposited and modified anodes
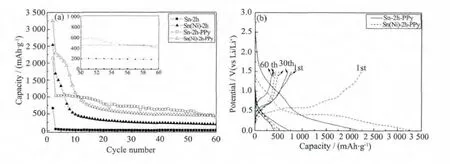
图6 沉积电极和修饰电极的循环寿命曲线(a)和充放电曲线(b)Fig.6 (a)Capacity vs.cycle number of deposited and modified anodes;(b)Potential vs.Capacity of deposited and modified anodes
图6(b)中首次充放电曲线显示Sn-2h-PPy首次放电比容量为2 149 mAh·g-1,首次库伦效率为35.3%,Sn(Ni)-2h-PPy首次放电比容量提高至3 252 mAh·g-1,首次库伦效率高达 68.8%,很明显,Sn(Ni)-2h-PPy首次库伦效率更高。分别比较两种材料的第30、60次充放电曲线,Sn(Ni)-2h-PPy电极的对应充放电曲线基本重合,而Sn-2h-PPy出现较大程度的衰减,说明Sn(Ni)-2h-PPy电极的循环稳定性更好。
聚吡咯位于沉积电极表面可作为保护膜减少电极表面活性物质与电解液的副反应,同时其良好的弹性可抑制体积效应引起的活性材料破碎,增加脱嵌锂过程可逆性,提高电极材料的循环稳定性。
图7为Sn(Ni)-2h-PPy电极的循环伏安曲线,是典型的Sn负极脱嵌锂过程。在还原曲线上位于0.65、0.75、0.8 V 的峰表示 Sn 的去合金过程,对应的合金化过程的峰出现在氧化曲线的 0.35、0.65 V,说明Sn(Ni)-2h-PPy电极的脱嵌锂过程是分步进行的[14-15]。没有对应于PPy脱嵌锂过程的峰出现,说明在0.05~1.5 V的电压范围PPy不参与脱嵌锂反应,对电极材料的容量没有贡献[16].
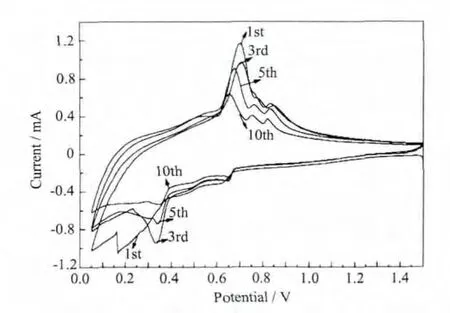
图7 Sn(Ni)-2h-PPy电极的循环伏安曲线Fig.7 Cyclic voltammogram of Sn(Ni)-2h-PPy electrode
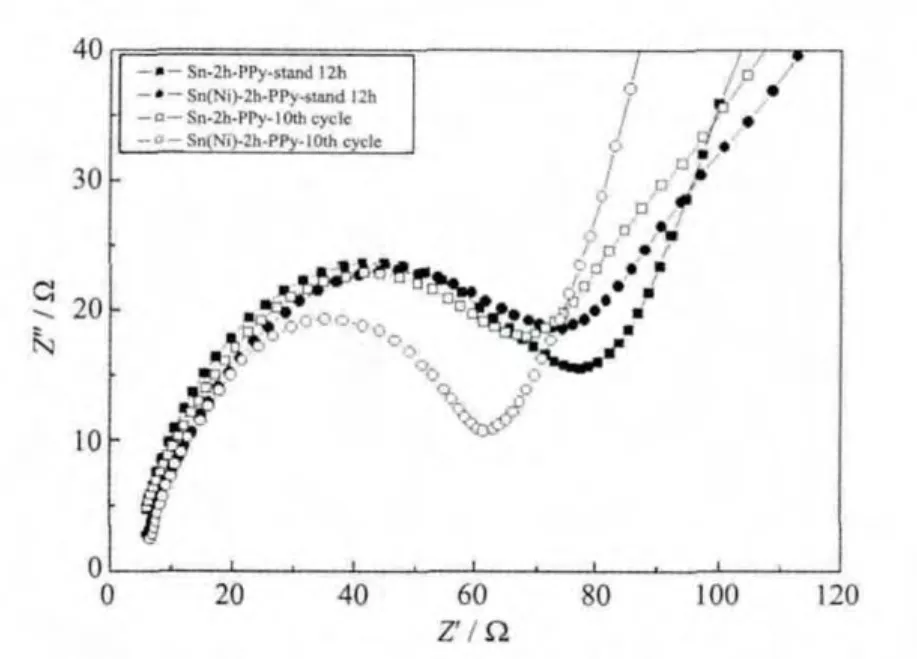
图8 修饰电极循环前后的阻抗谱Fig.8 EIS spectra of modified anodes before and after 10 cycles
图8为PPy修饰后电极循环前后的阻抗图。中高频区的半圆对应于电极的本征电子电阻和接触电阻以及金属锂对电极的表面钝化膜电阻[22-24],低频区斜线的斜率约为45°,代表Warburg阻抗[25]。循环后修饰电极在中高频区的阻抗都减小,说明电极经历了活化过程,开始展现优越的电化学性能。而Sn(Ni)-2h-PPy电极在经历10次循环后中高频区的阻抗明显比Sn-2h-PPy小得多,根据阻抗越大,则电导率越低,对应极材料的电化学性能越差,侧面证明了Sn(Ni)-2h-PPy电极的循环性能更稳定。
3 结 论
通过在铜箔上电沉积直接得到Sn负极,避开了粘结剂的使用。以NiCl2为沉积电解液的添加剂得到了Sn空心管,提高了材料的可逆比容量。进一步引入聚吡咯进行沉积电极表面修饰改性,有效地提高了电极的电化学循环稳定性,得到首次库伦效率高达 68.8%,60 次循环仍剩余 440.6 mAh·g-1可逆比容量的负极材料。
[1]Ng S H,Wang J Z,Wexler D,et al.Angew.Chem.Int.Ed.,2006,45(41):6896-6899
[2]Simon G K,Goswami T.Metall.Mater.Trans.,2011,42A:231-238
[3]Kim H,Han B,Choo J,et al.Angew.Chem.Int.Ed.,2008,47(52):10151-10154
[4]Tabuchi T,Hochgatterer N,Ogumi Z,et al.J.Power Sources,2009,188(2):552557
[5]Wolfenstine J,Campos S,Foster D,et al.J.Power Sources,2002,109:230-233
[6]Yang J,Takeda Y,Imanishi N,et al.Solid State Ionics,2000,135:175-180
[7]Zhao H L,Ng D H L,Lu Z Q,et al.J.Alloys Compd.,2005,395:192-200
[8]Wachtler M,Winter M,Besenhard J O.J.Power Sources,2002,105:151-160
[9]Cheng X Q,Shi P F.J.Alloys Compd.,2005,391:241-244
[10]Fang L,Chowdari B V R.J.Power Sources,2001,181:97-98
[11]Wang Y G,Li B,Zhang C L,et al.J.Power Sources,2012,219:89-93
[12]Veeraraghavan B,Paul J,Haran B,et al.J.Power Sources,2002,109:377-387
[13]Park K S,Schougaard S B,Goodenough J B.Adv.Mater.,2007,19(6):848-851
[14]Huggins R A.J.Power Sources,1999,81-82:13-19
[15]Zhang W M,Hu J S,Wan L J,et al.Adv.Mater.,2008,20:1160-1165
[16]Wang J,Too C O,Wallace G G,et al.J.Power Sources,2005,140:162-167
[22]Song J Y,Lee H H,Wang Y Y,et al.J.Power Sources,2002,111:255-267
[23]Zhang Y,Zhang X,Cheng H,et al.Electrochim.Acta,2006,51:4994-5000
[24]Jiang T,Zhang S,Qiu X,et al.Electrochem.Commun.,2007,9(5):930-934
[25]Dimov N,Kugino S,Yoshio M.Electrochim.Acta,2003,48(11):1579-1587
猜你喜欢
小读者·爱读写(2023年9期)2023-10-02 03:46:59
小读者(2023年18期)2023-09-27 04:38:38
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
山东冶金(2019年5期)2019-11-16 09:09:12
中国有色金属学报(2018年2期)2018-03-26 07:58:37
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
合成化学(2015年10期)2016-01-17 08:56:06
电源技术(2015年9期)2015-06-05 09:36:06
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
