
(顺)-1,1,1,4,4,4-六氟-2-丁烯的合成研究进展
2014-07-05 16:02赵波吕剑
化工进展 2014年1期
赵波,吕剑
(西安近代化学研究所,陕西 西安 710065)
(顺)-1,1,1,4,4,4-六氟-2-丁烯的合成研究进展
赵波,吕剑
(西安近代化学研究所,陕西 西安 710065)
作为氢氟烯烃(HFO)类物质,顺-1,1,1,4,4,4-六氟-2-丁烯(Z-HFO-1336)的臭氧消耗潜值(ODP)为零,温室效应潜值(GWP)极低,对环境影响很小,因其性能与前几代发泡剂相近,被看作新一代绿色环保发泡剂,其合成方法受到氟化工界的广泛关注。至今,Z-HFO-1336的合成方法主要可分为:①直接氟化C4化合物合成;②氟利昂类化合物CFC-113、HCFC-123经偶联反应制备;③由卤代甲烷与卤代烯烃经调聚反应制备。其中直接氟化法虽然路线短,但产物选择性低、催化剂稳定性差;而氟利昂类化合物偶联法污染大,调聚合成存在工艺复杂、收率低等问题。提出了两种具有学术和工业价值的研究路线:①HCFC-123自偶联一步合成Z-HFO-1336,反应涉及C—Cl键的活化、单电子转移(SET)等过程,极具理论研究意义;②以乙烯、三氟丙烯为原料经调聚反应合成,原料来源广泛,催化合成可连续进行,具有很强的工业价值。
(顺)-1,1,1,4,4,4-六氟-2-丁烯;发泡剂;合成方法
含氟烃类与聚醇基质具有良好兼容性和化学稳定性,以这类化合物为发泡剂制成的聚异氰酸酯、聚氨酯材料,具有低导热率,隔热性能优异,长期以来被用于家用电器和建筑材料等行业[1-2]。
由于第一、二代发泡剂(一氟三氯甲烷CFC-11、一氟二氯乙烷HCFC-141b等)破坏臭氧层,已经被禁止使用,而第三代发泡剂(1,1,1,3,3-五氟丙烷HFC-245fa等)会产生较强的温室效应[3-5]。随着全球变暖对地球生态影响的日益加剧,迫切需要寻找绿色环保的发泡剂。
顺-1,1,1,4,4,4-六氟-2-丁烯(Z-HFO-1336)因其分子中含有的双键,排入大气后很快便能与OH自由基加成,通过氧化而降解,大气存在寿命短(20天),温室效应值(GWP≈9)低。其沸点(32 ℃)与室温接近,发泡、隔热性能优异,成为最佳替代物之一[6-10]。
现有Z-HFO-1336合成方法可分为直接氟化、偶联转化和调聚合成3种,本文对现有制备方法进行评述,通过对比各类合成路线的优缺点,以期为设计新工艺提供有益的思路。
1 直接氟化
Z-HFO-1336的合成研究,早期集中于以C4化合物六氯丁二烯和丁烯二酸为原料,直接氟化合成,常用的氟化试剂包括HF、KF、SF4、F2、XeF2等[11-16]。
1948年,Henne首次报道了Z-HFO-1336的制备方法。先以三氟二氯化锑(SbF3Cl2)为催化剂,液相氟化六氯丁二烯合成2,3-二氯六氟-2-丁烯(CFC-1316),随后经锌粉脱氯,Raney-Ni加氢三步制备Z-HFO-1336[17-18],反应式见图1。但所采用的氟化试剂SbF3价格昂贵且用量较大,非均相制备催化剂耗时长,脱氯、加氢两步收率低(分别仅有54%和35%),留下了很大的改进空间。
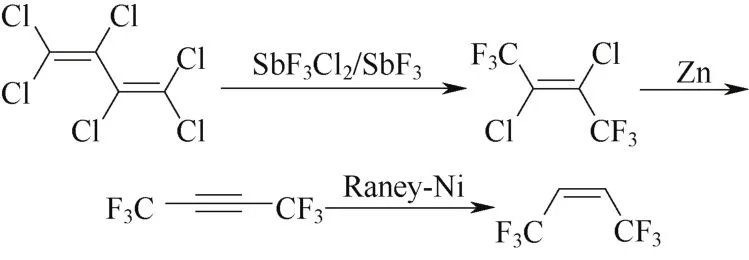
图1 方案1
Z-HFO-1336 也可经SF4氟化顺丁烯二酸一步合成[19],产物可保持与原料相同的反式构型,反应式见图2。但原料价格高, SF4气体毒性大,制备1 mol HFO-1336需消耗4 mol SF4,生成大量的SOF2、HF不仅原子利用率低,对环境污染也大。
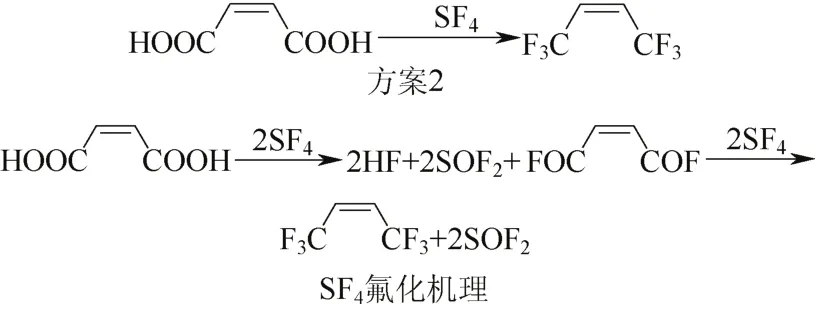
图2 方案2及反应机理
Maynard[20]以KF与六氯丁二烯在极性溶剂N-甲基-2-吡咯烷酮(NMP)中反应,意外的得到了加成了HF的产物七氟-2-丁烯,收率65%,七氟-2-丁烯可经脱氟化氢、加氢两步转化为HFO-1336。氟化仅在温度高于190 ℃时才能够进行,使用过量50%以上的KF,若工业应用,大量的废料难以得到有效处理。
1995年,Köln等[21]以五氯丁二烯为原料,经两次氟化法合成Z-HFO-1336。五氯丁二烯首先气相加成HF获得氟氯丁烷,再进行液相氟化。相比Maynard报道的工艺,KF用量降低了近50%。
Chambers等[22-23]利用全氟烃类(PFCs)十氟十氢菲作为反应介质,同样以KF氟化六氯丁二烯,将分离出的七氟-2-丁烯与六氟-2-丁炔混合物室温放置25天,产物全部转为六氟-2-丁炔,收率56%,反应式见图3。
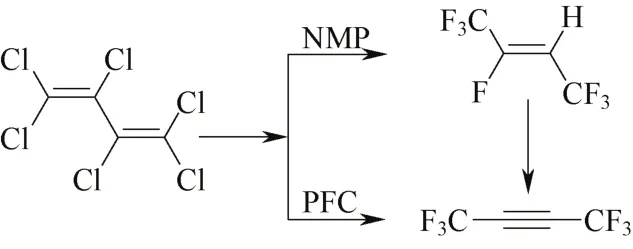
图3 方案3
直接氟化合成Z-HFO-1336的方法,需要消耗大量的氟化试剂,成本高,污染大,对此类工艺改进,应选择更廉价、高效的氟代试剂进行。
2 偶联转化
1990年前后,人们认识到氯氟烷烃CFCs和氢氯氟烷烃HCFCs类物质,一旦排入大气,产生的氯自由基会对臭氧层造成严重破坏,亟需寻找这类物质的降解途径[24-25]。降解可通过:氧化分解、加氢转化、偶联脱氯等途径[26-28]。
偶联反应作为有效降解氯原子的手段,不仅可将氯原子有效降解,生成的一些含氟烃类也可能成为新型发泡、制冷剂。
2.1 以CFC-113为原料
Johnson等[29]使用2%的Ru/C催化, 气相偶联氟利昂CFC-113a(1,1,1-三氯三氟乙烷)制备CFC-1316,在H2∶CFC-113a(摩尔比10∶1),反应温度125 ℃时选择性最佳(59%),但还原产物三氟乙烷含量近20%,一定程度上影响了收率,反应式见图4。
CFC-1316脱氯可通过金属脱氯和加氢脱氯:传统工艺以金属Zn在极性溶剂中脱氯,收率低、耗时长。近年报道的利用Fe(0)配合物的脱氯方法,室温即可进行,收率也提高至80%以上[30],表明CFC-1316脱氯以单电子转移(SET)进行更为有效。
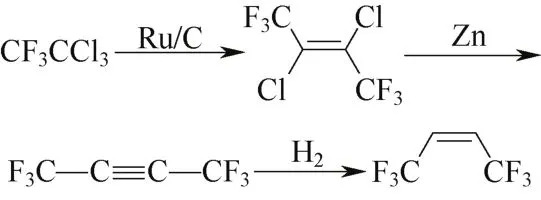
图4 方案4
Nappa等[31]选取多种金属及载体,对CFC-1316气相脱氯合成Z-HFO-1336进行了研究,发现在1%Cu/1%Ni/C催化下,反应温度375℃,H2/CFC-1316摩尔比7.5∶1,接触时间30 s时,HFO-1336的选择性能够达到80%以上,但以反式构型为主(75%左右)。
以六氟丁炔加氢合成Z-HFO-1336具有优异的顺式选择性,因此成为了近年的研究热点。Hazeldine[32]报道的以Raney-Ni催化,在15 atm(1 atm=101.325 kPa)、60 ℃下加氢,顺式产物收率91%,但分析发现产物含有少量六氟丁烷。六氟丁烷沸点(25 ℃左右)与Z-HFO-1336接近,不仅温室效应值高,而且会严重影响Z-HFO-1336的纯化。
Swearingen等[33-34]将六氟-2-丁炔室温加氢改以Lindlar Pd催化,通过增压(ΔP=50 psi,1 psi=6.895 kPa)控制反应进程,反式产物和六氟丁烷含量可控制在1%~2%,精馏后Z-HFO-1336收率59%。
Van Der Puy等[35]通过KOH、Pb等钝化Pd催化剂,并引入醇类稀释体系,室温加氢20 h,Z-HFO-1336的收率提高至97%左右。Jia等[36]则以0.1%的Bi钝化Pd/PAF,于150~200 ℃加氢,顺式产物选择性达到最佳90.2%。
六氟-2-丁炔加氢合成Z-HFO-1336能够达到较为理想的收率和选择性,但由于六氟-2-丁炔沸点较低(-25 ℃),作为中间体收集和储存都需要在低温操作,而选择以氯代丁烯直接加氢合成,虽然操作方便,但产物选择性和收率都有待进一步提高。
2.2 以HCFC-123为原料
1994年Aoyama等[37]在专利中报道,以金属铜与氟利昂HCFC-123(三氟二氯乙烷)为原料,通过二乙胺催化偶联合成Z/E-HFO-1336(收率44%),反应式见图5。Xu等[38]发现在Ullman反应中生成的还原产物三氟一氯乙烷(6%)难以分离,且仅在二乙胺作用下,反应才能获得相对理想的收率。Sun等[39]以2,2’-联吡啶为配体,在DMF中偶联HCFC-123,转化率提高至95%以上,避免了三氟一氯乙烷的生成。但由于反应中需使用化学计量比的Cu,用量较大,生产成本高。

图5 方案5
气相催化偶联收率及选择性不佳,而液相偶联需要消耗大量金属。随着CFCs和HCFCs类物质的停产,偶联路线原料来源受到很大影响,以廉价、来源更广的原料设计合成Z-HFO-1336成为设计的新思路。
3 调聚合成
多卤代烷与烯烃调聚合成新的C—C键(Kharasch反应),多半世纪以来,一直是有效增长碳链的方法。根据引发剂类型可分为:物理调聚、自由基调聚、催化调聚。早期使用物理调聚与自由基调聚,多聚产物较多,改由过渡金属催化(ATRA),机理见图6,1∶1调聚产物选择性大幅度提高[40-42]。
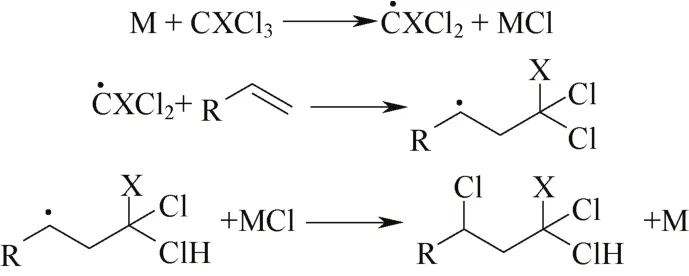
图6 ATRA机理
Hazeldine等[43]以三氟碘甲烷与1,1,1-三氟丙烯调聚,反应避光进行3天,合成2-碘六氟丁烷,经KOH脱HI后制得了E-HFO-1336,反应式见图7。但原料三氟碘甲烷没有理想的工业生产方法,研究只能停留在实验室阶段。
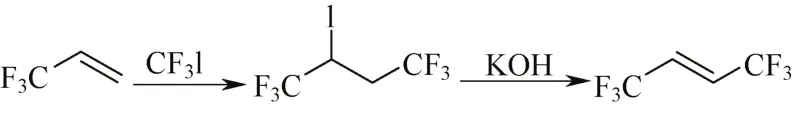
图7 方案6
Nappa[44]以四氟乙烯(TFE)和二氯四氟乙烷(CFC-114a)为原料,先通过氟氯化铝(AlClxF3-x)催化调聚制备2,2-二氯八氟丁烷CFC-318ma,气相进行Ni/Cu/Cr/CaF2催化脱卤,最后经Lindlar Pd催化加氢分4步合成Z-HFO-1336,反应式见图8。但连续进行的两次脱卤转化率低,二次脱卤选择性甚至不及20%,对收率影响很大。
Tung等[45]于2011年连续发表了两篇调聚合成Z-HFO-1336的专利。通过Fe/FeCl3/ Bu3PO4催化调聚三氟丙烯与四氯化碳制备四氯三氟丁烷,产物为以Cr基催化剂气相氟化或以SbCl5催化液相催化氟化合成Z-HFO-1336,反应式见图9。
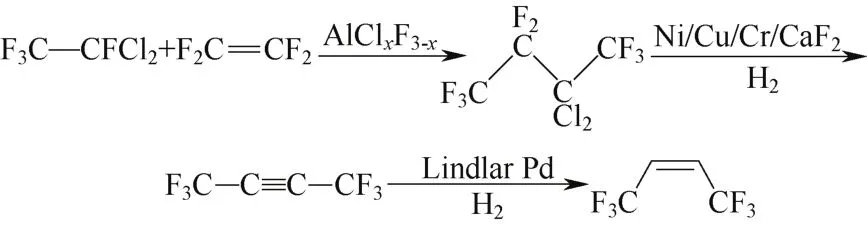
图8 方案7
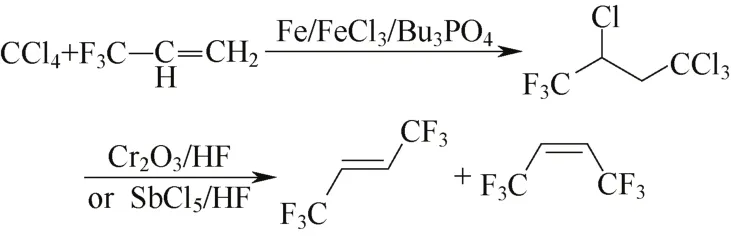
图9 方案8a
另一路线则以更为廉价的乙烯与四氯化碳为原料,通过Fe/FeCl3/Bu3PO4催化进行1∶2调聚,制备七氯丁烷,同样经氟化合成Z-HFO-1336[46-47],反应式见图10。
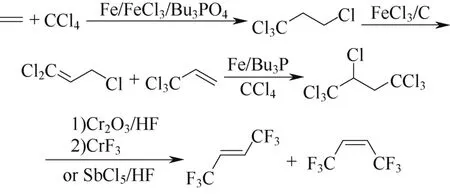
图10 方案8b
需要指出的是经脱卤化氢合成Z-HFO-1336,以反式消除为主,造成产物中反式比例较大,为此Tung以CrF3催化转型E-HFO-1336,250 ℃、1 atm下转化率61%。
Poss等[48-51]于2011年设计了3条调聚合成Z-HFO-1336的路线:以全氟丙烯与卤代甲烷为原料合成Z-HFO-1336,首先气相热聚制备三氯六氟丁烷,产物通过液相氟化,再经Ni-Cu-Cr催化脱氟、加氢合成Z-HFO-1336,反应式见图11。由于全氟烃类性质稳定,脱氟的收率是该路线的关键。
以四氯化碳与二氯三氟丙烯为原料合成Z-HFO-1336,调聚在CuCl2催化下,130~140 ℃进行16 h,产物可通过液相(SbCl5催化)或气相氟化(Cr2O3/Al2O3催化)制备六氟氯代丁烷,经脱氯、加氢合成Z-HFO-1336,反应式见图12。该路线不仅路线长,脱氯时需使用大量强碱和金属,成本明显偏高。

图11 方案9a
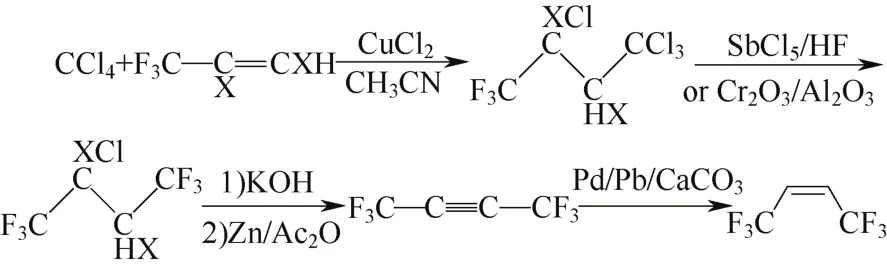
图12 方案9b
以CFC-113a和氯代乙烯为原料的调聚在Fe/ Bu3PO3催化下进行,氯化该调聚产物制备七氯三氟丁烷,再液相氟化、Cu/C催化脱氯合成Z-HFO-1336,反应式见图13。作为原料的CFC-113a可由四氯乙烯经液相氟化合成,但脱氯时顺式选择性不高等问题限制了该路线的工业级应用。
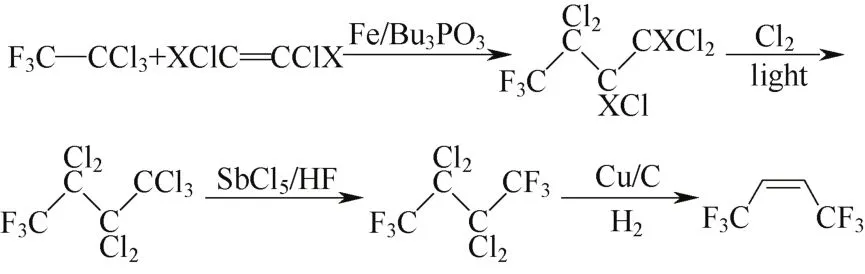
图13 方案9c
虽然多卤代烷和烯烃的调聚工艺成熟,但用以合成Z-HFO-1336,路线长且操作复杂,若简化步骤,选择性也随之降低,设计相关调聚工艺,需综合考虑工艺繁简度与选择性的关系。
4 结 语
现有相关合成Z-HFO-1336的文献报道,以改进和提高顺式产物的工艺和收率研究为主。直接氟化方法需消耗大量的氟化剂,成本高,污染大;偶联路线选取的起始物料已被《蒙特利尔协议》所禁止生产,不再廉价易得;而以小分子调聚合成的路线工艺复杂且步骤冗长,这些都是设计新工艺时亟待解决的问题。
液相偶联HCFC-123合成Z-HFO-1336,步骤短,转化率高(97%以上),但产物中反式含量较大(80%左右),以提高顺式产物含量的改进方法极具学术价值;另一方面,改进乙烯或三氟丙烯与四氯化碳调聚合成Z-HFO-1336的路线,由于原料来源便捷,工艺中所需的氟化、脱氯等工艺成熟和环保具有很强的工业应用前景。
[1] Baasandorj M,Ravishankara A R,Burkholder J B. Atmospheric chemistry of (Z)-CF3CH=CHCF3:OH radical reaction rate coefficient and global warming potential[J]. J. Phys. Chem. A,2011,115(38):10539-10549.
[2] Harvey L D D. Net climatic impact of solid foam insulation produced with halocarbon and non-halocarbon blowing agents[J]. Building and Environment,2007,42:2860-2879.
[3] Hedrick V,Chien J,Boggs J,et al. Fire extinguishing and fire suppression compositions comprising unsaturate fluorocarbons:WO,20070059468[P]. 2007-5-24.
[4] Robin M L. Azeotropic and azeotrope-like compositions of E-,1,1,1,4,4,4-hexafluoro-2-butene:WO,2008154612[P]. 2008-12-18 [5] Loh G,Creazoo,J A. Compositions and use of trans-1,1,1,4,4,4-hexafluoro-2-butene foam-forming composition in the preparation of polyisocyanate-based foams:WO,2009014966[P]. 2009-1-29.
[6] Eckert E R G,Goldstein R J,Ibele W E,et al. Heat transfer-a review of 1993 literature[J]. Int. J. Heat Mass Transfer,1996,39(5):885-963.
[7] Antunes M,Velasco J I,Solorzano E,et al. Heat transfer in polyolefin foams,in heat transfer in multi-phase materials[M]. Berlin Heidelberg:Springer,2011:131-161.
[8] Föglein K A,Szépvölgyi J,Szabó P T,et al. Comparative study on decomposition of CFCl3in thermal and cold plasma[J]. Plasma Chemistry and Plasma Processing,2005,25(3):275-288.
[9] 付庆,孙晓刚,向绍基,等.六氟-2-丁烯的合成研究进展[J]. 有机氟工业,2012(1):30-33.
[10] 吕涛.新型聚氨酯发泡剂1,1,1,4,4,4-六氟丁烯[J]. 有机氟工业,2012(2):29-33.
[11] Hutchinson J,Sandford G. Organoflurine chemistry:Element fluorine in organic chemistry[J]. Top. Curr. Chem.,1997,193:1-43.
[12] Wilkinson J A. Recent advances in the selective formation of the CF Bond[J]. Chem. Rev.,1992,92:505-519.
[13] Barton D H R,Godinho L S,Hesse R H,et al. Organic reactions of fluoroxy-compounds:Electrophilic fluorination of activated olefins[J]. Chem. Commun.,1968(14):804-808.
[14] Schack C J,Christe K O. Reactions of fluorine perchlorate with fluorocarbons and the polarity of the O-F bond in covalent hypofluorites[J]. Inorg. Chem.,1979,18(9):2619-2620.
[15] Tius M A. Xenon difluoride in synthesis[J]. Tetrahedron,1995,51 (24):6605-6634.
[16] Schmutzler R. Nitrogen oxide fluorides[J]. Angew. Chem. Int. Ed. Engl.,1968,7(6):440-455.
[17] Henne A L,Finnegan W G. Perfluoro-2-butyne and its hydrogenation products[J]. J. Am. Chem. Soc.,1949,71:298-300.
[18] Henne A L,Newman M S,Quill L L,et al. The alkaline condensation of fluorinated esters with esters and ketones[J]. J. Am. Chem. Soc.,1947,69:1819-1820.
[19] Hasek W R,Smith W C,Engelhardt V A. The chemistry of sulfur tetrafluoride. Ⅱ. the fluorination of organic carbonyl compounds[J]. J. Am. Chem. Soc.,1960,82:543-551.
[20] Maynard J T. The synthesis of highly fluorinated compounds by use of potassium fluoride in polar solvents[J]. J. Org. Chem.,1963,28:112-115.
[21] Köln N L,Leverkusen A M,Ratingen D B. Process for preparing hexafluorobutene:US,19955463150[P]. 1995-10-31.
[22] Chambers R D,Roche A J. Eliminations from 2H-Heptafluorobut-2-ene[J]. Journal of Fluorine Chemistry,1996,79:121-124.
[23] Chambers R D,Edwards A R. Perfluorocarbon fluids as solvent replacements[J]. J. Chem. Soc. Perkin Trans.,1997(24):3623-3627.
[24] Rowland F S. Stratospheric ozone depletion[J]. Phil. Trans. R. Soc. B,2006,361:769-790.
[25] Kim K H,Shon Z H,Nguyen H T,et al. A review of major chlorofluorocarbons and their halocarbon alternatives in the air[J]. Atmospheric Environment,2011,45:1369-1382.
[26] Liang C W,Ku C K,Chen Y C,et al. The performance of Ba in total oxidation of chlorinated hydrocarbons over La-Ba-Ni mixed oxide catalysts[J]. Cat. Comm.,2012,17:43-48.
[27] Makkee M,van de Sandt E J A X,Wiersma A,et al. Development of a satisfactory palladium on activated carbon catalyst for the selective hydrogenolysis of CCl2F2(CFC-12) into CH2F2(HFC-32)[J]. Journal of Molecular Catalysis A,1998,134:191-200.
[28] Ma Z,Hua W M,Tang Y,et al. Catalytic decomposition of CFC-12 over solid acids WO3/MxOy(M=Ti,Sn,Fe)[J]. Journal of Molecular Catalysis A,2000,159:335-345.
[29] Johnson R W,Moeller W C,Van De Puy M,et al. Process for combining chlorine-containing molecules to synthesize fluorinecontaining products:WO,9505353[P]. 1995-2-23.
[30] Thoreson K A,McNeill K. Vicinal dichlorine elimination at dichloroalkenes promoted by a well-defined iron(0) complex[J]. Dalton Trans.,2011,40:1646-1648.
[31] Nappa M J,Swearingen E N. Method of hydrodechlorination to produce dihydrofluorinated olefins:WO,2009006358[P]. 2009-1-8.
[32] Haszeldine R N. The Addition of free radicals to unsaturated systems. Part Ⅰ. The direction of radical addition to 3∶3∶3-trifluoropropene [J]. J. Chem. Soc.,1952:2504-2513.
[33] Swearingen E N. High selectivity process to make dihydrofluoroalkenes:US,20080269532[P]. 2008-10-30.
[34] Swearingen E N. High selectivity process to make dihydrofluoroalkenes:WO,2009142642[P]. 2009-11-26.
[35] Van Der Puy M,Ma J J. Process for preparation of perfluorinated cis-alkene:WO,2010014548[P]. 2010-2-4.
[36] Jia X Q,Zhou X M,Quan H D,et al. Preparation of cis-1,1,1,4,4,4-hexafluorobut-2-ene by cis-selective semihydrogenation of perfluoro-2-butyne[J]. Journal of Fluorine Chemistry,2011,132:1188-1193.
[37] Nakada T,Aoyama H,Takubo S. Process for producing 1,1,1,4,4,4-hexafluorobutene and 1,1,1,4,4,4-hexafluoro-butane:WO,9417020[P]. 1994-8-4.
[38] Xu Y L,Dolbier W R,Rong X X. 1,1-Bis (dimethylamino)-2,2,2-trifluoroethane,a readily-available precursor to the novel fluorinated building block 1,1-bis(dimethylamino)-2,2-difluoroethene [J]. J. Org. Chem.,1997,62:1576-1577.
[39] Sun X H,Nappa M J,Lee W C. Process for making 1,1,1,4,4,4-hexafluoro-2-butene:WO,2009117458[P]. 2009-9-24.
[40] Minisci F. Free-radical additions to olefins in the presence of redox systems[J]. Acc. Chem. Res.,1975,8(5):165-171.
[41] Iqbal J,Bhatia B,Nayyar N K. Transition metal-promoted free-radical reactions in organic synthesis:The formation of carbon-carbon bonds[J]. Chem. Rev.,1994,94:519-564.
[42] Gossage R A,Van de Kuil L A,Van Koten G. Diaminoarylnickel(II) “pincer” complexes:Mechanistic considerations in the kharasch addition reaction,controlled polymerization,and dendrimeric transition metal catalysts[J]. Acc. Chem. Res.,1998,31:423-431.
[43] Leedham K,Haszeldine R N. The direction of radical- addition to alkyl- and perjluoroalkyl-acetylenes[J]. J. Chem. Soc.,1954: 1634-1638.
[44] Nappa M J. Process for the synthesis of 2-chloro- 1,1,1,3,3,4,4,4-heptafluoro-2-butene and hexafluoro-2-butyne:US,20090156869[P]. 2009-06-18.
[45] Tung H S,Wang H. Method for making hexafluoro-2-butene:WO,2011119370[P]. 2011-09-29.
[46] Tung H S,Wang H. Method for manufacture of hexafluoro-2-butene:WO,2011119388[P]. 2011-09-29.
[47] Nappa M J,Swearingen E N. Catalytic synthesis of internal fluorobutenes and internal fluoropentenes:WO,2012067864[P]. 2012-05-24.
[48] Poss A J,Nalewajek D,Nair H K,et al. Process for cis 1,1,1,4,4,4-hexafluoro-2-butenes:US,20110288346[P]. 2011-11-24.
[49] Poss A J,Nalewajek D,Nair H K,et al. Process for cis 1,1,1,4,4,4-hexafluoro-2-butenes:WO,2011146820[P]. 2011-11-24.
[50] Poss A J,Van Der Puy M,Singh R R,et al. Process for the preparation of fluorinated cis-alkene:WO,2011146792[P]. 2011-11-24.
[51] Poss A J,Nalewajek D,Van Der Puy M,et al. Process for the production of fluorinated cis-alkene:WO,2011146802[P]. 2011-11-24.
Progress of preparation of cis-hexafluoro-2-butene
ZHAO Bo,LÜ Jian
(Xi’an Modern Chemistry Research Institute,Xi’an 710065,Shaanxi,China)
Hydrofluoroolefin cis-1,1,1,4,4,4-hexafluoro-2-butene (Z-HFO-1336) has zero ozone depletion potential (ODP) and low global warming potential (GWP),which replaces trichlorofluoromethane CFC-11,1,1-dichloro-1-fluoroethane HCFC-141b,and 1,1,1,3,3-pentafluoropropane HFC-245fa by good compatibility and chemical stability,is used as new generation foaming materials. Synthesis of the compound mainly includes three types:direct fluorination of C4compounds,coupling of CFC-113 and HCFC-123,and telomerization of haloalkane with olefin to form chlorofluorobutane by Kharasch reaction. Direct method is too expensive for large scale production,coupling route brings up pollution problems,and complicated Kharasch reaction involves long steps. Two synthesis approaches are proposed:Coupling of HCFC-123 attracts theoretical study,which involves activation of C─Cl bond and single electron transfer process,and simple and practical way using ethylene or trifluoropropene as starting material could be an effective method for plant-scale production.
cis-1,1,1,4,4,4-hexafluoro-2-butene;blowing agent;synthetic
TQ 222.4
A
1000-6613(2014)01-0193-06
10.3969/j.issn.1000-6613.2014.01.034
2013-08-05;修改稿日期:2013-08-25。
赵波(1983—),男,博士研究生。联系人:吕剑,研究员,博士生导师。E-mail lujian204@263.net。
猜你喜欢
有机氟工业(2020年2期)2020-07-04
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
石油石化绿色低碳(2019年6期)2019-01-14
合成化学(2015年10期)2016-01-17
合成化学(2015年10期)2016-01-17
化学反应工程与工艺(2015年3期)2015-04-16
应用化工(2014年1期)2014-08-16
有机氟工业(2014年3期)2014-06-05
影像科学与光化学(2014年5期)2014-03-11
中国医药科学(2013年16期)2013-12-20
