
互通多孔碳/二氧化锰纳米复合材料的原位水热合成及电化学性能
2014-06-23 06:51:56张宣宣范会利孔令斌
物理化学学报 2014年5期
张宣宣 冉 奋,2,* 范会利 孔令斌,2 康 龙,2,*
(1兰州理工大学材料科学与工程学院,兰州730050;2兰州理工大学甘肃省部共建有色金属先进加工与再利用国家重点实验室,兰州730050)
1 引言
超级电容器是一种新型的绿色储能器件,其储能性能介于二次电池和传统电容器之间.1它具有比功率高、比容量大、成本低、循环寿命长、充放电效率高等优点,因此在移动通讯、信息技术、电动汽车、航空航天和国防科技等领域具有广阔的应用.超级电容器研究核心是电极材料,一般分为:炭材料、过渡金属氧化物和导电聚合物.2,3炭材料研究历史最长,技术最成熟,商业化程度最高,主要提供的是双电层电容,但其比容量相对较低,能量密度较小;相比较而言金属氧化物和导电聚合物提供的是法拉第赝电容,比容量较大、能量密度较高,但其导电性能较差,循环稳定性不好.4,5
为了协同和弥补这些材料性能之间的优缺点,已经有很多诸如碳基材料与碳基材料、碳基材料与过渡金属氧化物及碳基材料与导电聚合物等的复合材料被应用于超级电容器领域,而制备融合双电层电容和赝电容优势的复合材料已成为电极材料研究的热点.在赝电容材料中,二氧化锰(MnO2)由于在自然界中含量丰富、价格低廉、高的理论比容量(700-1380 F·g-1),且对环境友好,6,7被认为是最有前景的电极材料.但是,MnO2的导电性较差且发生法拉第赝电容反应是表面反应,即仅在表面或表面很薄的一层才能发生赝电容反应,这使得MnO2的实际比容量仅有120-250 F·g-1.8将MnO2复合在具有大的比表面积的碳基材料上(如炭黑、9碳片状纸、10石墨烯、11-13碳纳米管14-17和介孔碳18)已有大量的研究.利用碳材料的大比表面积和优良的导电性能,分散MnO2提高其电容利用率.碳和MnO2复合材料常用的制备方法有微波辐射法、溶胶-凝胶法、电化学沉积法、微乳液合成法、共沉淀法、化学沉淀法和水热法等.13-23
本文采用了简单快速的水热法合成了互通多孔碳/二氧化锰纳米片状复合电极材料,选用了简单易得,具有大的比表面积及丰富孔结构的IPC作为碳骨架,并且利用IPC和高锰酸钾之间可逆的氧化还原反应方程式(1):24
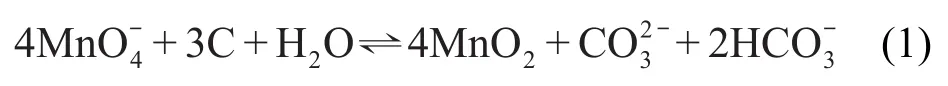
使得原位生成的MnO2均匀地负载在IPC表面.由于IPC和MnO2大的接触面积及IPC良好的导电性,使得表面负载的MnO2可以充分发生赝电容反应.同时,研究了反应温度对表面负载的MnO2形貌和复合材料的电化学性能影响.最后,组装成IPC/MnO2//AC混合超级电容器,研究其电化学电容特性及大电流充放电性能.
2 实验部分
2.1 药品
苯乙烯(St,分析纯),国药集团化学试剂有限公司;二乙烯基苯(DVB,分析纯),J&K百灵威;S-pan80(分析纯),中国莱阳双双化工有限公司;甲苯(T,分析纯),过硫酸钾(K2S2O8,分析纯)、无水氯化钙(CaCl2,分析纯)及高锰酸钾(KMnO4,分析纯),天津市德恩化学试剂有限公司.
2.2 IPC的制备
首先,超浓乳液聚合合成互通多孔聚苯乙烯材料(Interconnected Porous Polystyrene,简记为IP.PS),25配置连续相(4 mL St、4 mL DVB、5 mL T 和 3.0 g Span80)加入带有电动搅拌器和温度计的三口烧瓶,室温搅拌形成均相溶液.然后通过滴液漏斗滴加分散相溶液(蒸馏水85 mL、0.2 g K2S2O8和l.0 g无水CaCl2),约30 min滴加完毕,并控制温度为55°C,滴加完毕后继续反应10 min,最后转移到60°C的水浴锅中,反应48 h,得到的白色块状物质即为制备的IP.PS.再将所得的白色块状物IP.PS在马弗炉中进行5 h热处理,热处理的温度为320°C,主要是为了让聚苯乙烯更好地交联.然后将样品在管式炉中在700°C,N2氛围中处理2 h,25即可得到互通多孔碳材料.
互通多孔碳材料的后期酸化处理,将IPC材料置于80°C的浓硝酸中回流4 h,然后进行抽滤洗涤至滤液的pH值接近7,得到改性互通多孔碳材料.
2.3 IPC/MnO2复合电极材料的制备
称取100.0 mg改性的IPC置于300 mL的去离子水中,超声分散30 min,再加入250.0 mg的KMnO4超声10 min后移入水浴锅中磁力搅拌反应6 h,用蒸馏水、无水乙醇各清洗3遍后烘干得到IPC/MnO2复合电极材料.
2.4 IPC/MnO2复合电极材料的结构及性能测试
本实验采用粉末X射线衍射(XRD,D/Max-2400,日本Rigaku公司,CuKα,λ=0.154178 nm)分析其物相结构.采用场发射电子扫描电镜(SEM,JSM-6701F,日本JOEL公司)和透射电镜(TEM,JEM-2010,日本JEOL公司)观察材料的形貌.采用氮气吸附-脱附仪(ASAP 2020,美国Micromeritics公司)来测定电极材料在77 K下的N2吸附等温线,用BET、BJH法分别计算电极材料的比表面积和孔径分布;采用同步热分析仪(DSC/DTA-TG,STA 449 F3,德国NETZSCH公司)进行热重分析(TGA),在空气氛围中,以升温速率为10°C·min-1在25-900°C范围内进行测试.采用电化学工作站(CHI660C,上海辰华仪器公司)和LAND电池测试系统(CT2001A,武汉市蓝电电子有限公司)在三电极体系测试工作电极的电化学性能.饱和甘汞电极为参比电极,铂电极为对电极,电解质为1 mol·L-1Na2SO4溶液,工作电极的几何面积为1 cm×1 cm,电位窗口为0-1 V.循环伏安主要测试不同扫描速率下的循环伏安特性曲线.恒流充放电主要测试工作电极在不同的电流密度下的充放电曲线及其功率特性.电极比电容计算采用公式(2)和(3):

其中,Cm(F·g-1)为比容量,i(A)为放电电流,Δt(s)为放电时间,ΔV(V)为放电电压降,s(V·s-1)为扫描速率,m(g)为电极活性物质的质量.26,27
2.5 IPC/MnO2//AC混合超级电容器的组装和性能测试
混合超级电容器由两个独立电极组成,IPC/MnO2作为正极材料,AC作为负极材料(所用商业活性碳购于邵武鑫森碳业公司未经任何处理,AC的比表面积为2000 m2·g-1),两电极用电解液可透过的多孔隔膜隔开,电解质为1 mol·L-1Na2SO4溶液,单电极的几何面积为1.5 cm2,电位窗口为0-1.8 V.
超级电容器遵循电荷平衡q-=q+(这里q-和q+分别表示正极和负极存储的电荷量).单电极存储的电荷量和每个电极材料的比容量C、电位窗口ΔE(V)及电极材料的质量m(g)有关,电荷量的计算采用公式(4):28

由q-=q+,所以可以得到正极材料m-(g)与负极材料m+(g)的质量比的公式(5):28
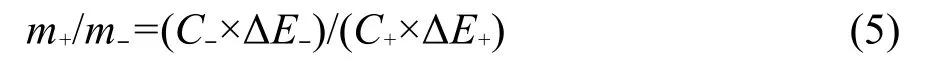
三电极体系测得活性炭和IPC/MnO2复合电极材料的比容量分别为162和173 F·g-1.根据活性炭和IPC/MnO2单电极比容量和电位窗口的大小,可以计算得到混合电容器中正负极材料的质量比m+/m-=0.93.因此,活性物质和IPC/MnO2材料的质量分别为16.6和8.0 mg.采用电化学工作站对混合电容器进行电化学性能测试.
3 实验结果与讨论
3.1 材料的制备及结构分析
IPC/MnO2复合材料合成机理图如图1所示,基于KMnO4与IPC之间的可逆反应采用水热法原位合成IPC/MnO2复合电极材料.反应方程式如式(1)所示,在这个反应中,IPC既作为骨架又作为还原剂.KMnO4首先与IPC表面的碳接触,碳将KMnO4还原成MnO2并均匀地沉积在IPC表面,同时表面的碳被刻蚀掉.随着反应的进行,体系中KMnO4的浓度逐渐降低,水热反应的速率也逐渐降低.碳与KMnO4的进一步反应生成更多的MnO2,生成的MnO2在载体表面形成纳米片状结构.由于MnO2纳米片状的生长和碳表面的刻蚀是同时进行的,因此在碳表面形成了多层次的纳米空隙结构.另外,所选择碳骨架本身就是具有互通的多孔结构,使得制备的IPC/MnO2具有非常丰富的孔结构和纳米结构,这非常有利于电解液与电极接触的动力学过程,并提供良好的电解液缓冲基地.
图2a是互通多孔碳载体的扫描电镜图,由图可以看出IPC呈蜂窝状骨架结构,表面有大小不一的孔存在,碳骨架结构的孔径约为10-20 μm,骨架的表层孔的孔径分布约在2-10 μm之间.图2(b,c,d)分别为温度在30、60及100°C时制备IPC/MnO2复合材料的扫描电镜图.在30°C时MnO2以纳米颗粒堆积成三维孔结构覆盖在IPC表面,颗粒的尺寸约在100 nm.在60°C时MnO2以纳米片及由纳米片卷曲成的锥形纳米棒覆盖在IPC表面,棒长约200 nm,直径约40 nm.在温度为100°C时由纳米棒堆积的三维网状结构包覆在IPC表面,棒直径约为30 nm.60和100°C时制备的MnO2形貌非常相似,在100°C时纳米片几乎都卷曲呈锥刺状结构的存在.在温度较低时,碳的刻蚀速率较慢,所以锰源在水热条件下以较低速率生长出纳米颗粒晶核,随着水热反应的进行,纳米颗粒开始变得不稳定,呈现出团聚趋势.随着温度升高,锰氧八面体基本结构单元在水热条件下浓缩成薄片状的MnO2.当体系温度不断升高,压力不断增大时,薄片状MnO2呈现出卷曲趋势,溶液中出现部分层状结构的MnO2,这是一种亚稳态结构.当溶液中存在足量阳离子作稳定剂时,层状结构才能保持原状;因为溶液中存在适量K+,层状结构MnO2直接坍塌为隧道型MnO2.且温度越高纳米片的厚度越薄而更容易发生卷曲,这样会使得其比表面积增大.29,30由此可见,温度对MnO2形貌的影响较大,相比之下高温下(100°C)得到的结构更适合电解液离子的传输扩散,从而使得高温下的复合材料具有良好的电化学电容性能.
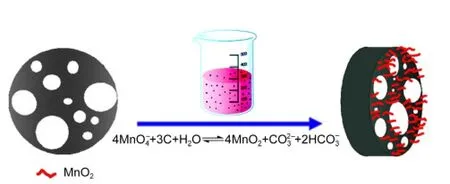
图1 IPC/MnO2复合材料的合成机理图Fig.1 Procedure for preparation of IPC/MnO2composite

图2 IPC和不同温度下制备的IPC/MnO2复合材料的SEM图Fig.2 SEM images of the IPC and the IPC/MnO2composites prepared at different reaction temperatures
图3为100°C时制备的IPC/MnO2复合材料的TEM照片.从图可以看出,(1)碳材料呈现多孔结构,具有100 nm以下的多尺度孔径分布;(2)MnO2呈纳米级结构,均匀地负载在IPC表面,负载厚度约有150 nm.
由于制备过程中利用碳骨架和高锰酸钾反应,碳骨架在其中起还原高锰酸解的作用.经过反应后碳骨架被刻蚀,且产生纳米结构的MnO2包覆在碳骨架上形成新的孔隙结构,那么反应前后材料的孔结构和比表面积会发生变化.图4为IPC和IPC/MnO2的N2吸脱附等温线,插图为各自样品的孔径分布图.图中滞回线表明材料都呈现多孔性固体的吸附特征,IPC和IPC/MnO2比表面积分别是280和316 m2·g-1,BJH平均脱附孔径分别是25和50 nm.从IPC孔径分布曲线可以看出其孔径的尺寸具有<2 nm的微孔结构,还有2-5 nm、6-15 nm的介孔结构.而从IPC/MnO2孔径分布曲线可以看出,孔结构主要以孔径尺寸为3-4 nm、4-15 nm、15-20 nm的介孔结构存在.结果表明碳骨架上<2 nm的微孔结构及小介孔经刻蚀之后孔径有所增大,且由于二氧化锰片堆积卷曲包覆在IPC形成新的介孔结构,从而增加了复合材料比表面积和BJH平均脱附孔径.
由图5(a)可知,IPC/MnO2复合电极材料在12°、24°、37°及66°有明显的特征峰,对比K-Birnessite型MnO2的标准谱JCPDS 52-0556可知,这些峰均为K-Birnessite型MnO2的特征峰,化合物的分子式为K0.27MnO2(H2O)0.54.图5(b)为IPC/MnO2复合材料在25-900°C范围内的热重曲线,在温度150°C之前有明显的失重峰,主要是结合水的失去;在200-500°C之间主要是互通多孔碳的烧损,在此过程中并伴有锰形态的转化(即MnO2→1/2 Mn2O3);在500-900°C形成了Mn3O4.31所以,当温度达到900°C,剩下的只有锰的氧化物Mn3O4.依据原子质量守恒及元素守恒定律,由Mn3O4→Mn2O3→MnO2的变化计算可得IPC/MnO2复合材料中MnO2的含量约为34%.

图3 IPC/MnO2复合材料的TEM照片Fig.3 TEM image of the IPC/MnO2composite
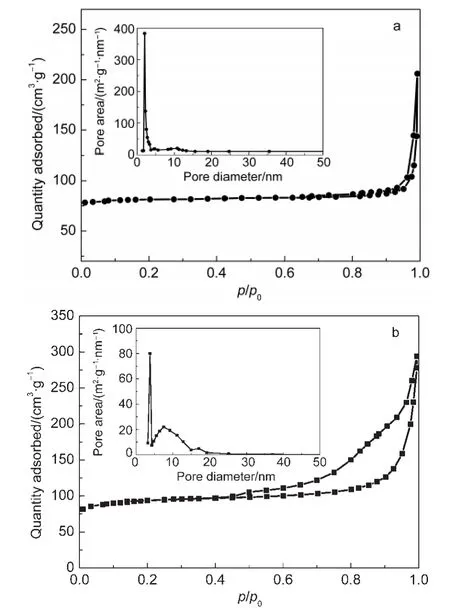
图4 氮气吸脱附曲线Fig.4 N2adsorption/adsorption isotherms
3.2 IPC/MnO2复合电极材料电化学性能
通过对MnO2的充放电机理研究,大多数科学家都认为质子-电子机理是比较正确的反应机理.质子-电子机理认为MnO2晶格是Mn4+与O2-交错排列而成的,反应过程是液相中的质子通过两相界面进入MnO2晶格中与O2-结合为OH-,而电子也由外部进入锰原子的周围使四价锰还原为低价锰.原来O2-晶格点阵被OH-代替,形成水锰石MnOOH,这是MnO2还原的初级产物.在中性溶液中,反应为:32,33

MnO2还原的初级反应产物存在于MnO2颗粒表面上,当它与电解液进一步发生化学反应或通过其它方式离开电极表面的过程,称为次级反应.当pH值较低时,水锰石的转移通过下述反应进行:
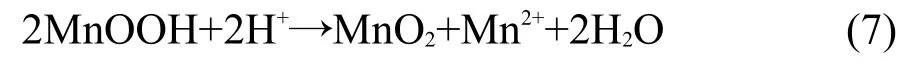
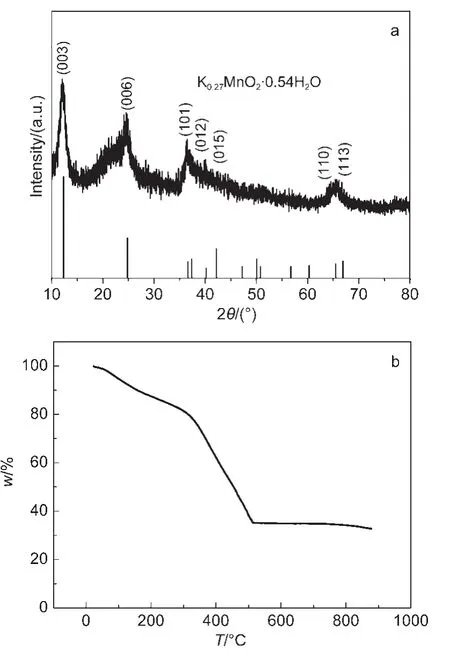
图5 IPC/MnO2复合材料的(a)XRD和(b)TGA曲线Fig.5 (a)XRD and(b)TGAcurves of the IPC/MnO2 composite
充电时,MnO2吸附溶液中的H+,同时接受电子,被还原为MnOOH,放电时,MnOOH失去电子,并释放出H+,被氧化为MnO2.这样电极便将电荷储存释放出来,从而表现出法拉第电容的性质.由图6A可知,在低扫描速率下,曲线呈良好的矩形,随着扫描速率的增加,曲线从类矩形到柳叶状.由此可见增大扫描速度后,循环伏安曲线开始变形,充放电电流响应也随扫描速率的增加而增大,标志着快速的电流-电压响应.根据比容量计算公式(3)可以计算得知,扫描速率从5 mV·s-1到500 mV·s-1,增加了100倍,则MnO2比容量从897 F·g-1减小到22 F·g-1.图6B中曲线分别是在温度为30、60和100°C时循环伏安曲线.由图可见,不同温度制备的电极材料的循环伏安曲线都没有明显的氧化还原峰,呈比较对称的矩形,说明在扫描电位范围内,氧化还原反应均匀地进行,其中100°C曲线的对称性较好,说明在电化学反应过程中具有良好的可逆性和较好的电化学性能.
由图7A可以看出100°C制备的IPC/MnO2复合材料在电流密度为5 mA·cm-2时基本呈对称的三角形,说明电极反应具有良好的可逆性,其比容量为411 F·g-1(由于在中性及碱性电解液中碳的电位窗口为-1-0 V,所以在-1-0 V的电压范围内忽略互通多孔炭对容量贡献).随着电流密度的增大,电势降IR逐渐增大,比容量也有所下降.造成复合电极材料在较大电流下电容性能下降的原因可能有两点:一是由于在大电流充放电时,电极上短时间内吸附大量H+,电极/电解液界面上的H+浓度的急速下降,而H+的扩散速度相对较慢,界面上的H+的数目并不能满足电极充电所需的浓度,这样造成电极上由液相扩散引起的极化增大,并逐渐成为控制步骤,所以外加电势虽然不断上升.但是在电极上充入的电荷却没有相应的增加速度,从而引起电极在大电流下的容量损失.二是当电极在较大的电流下工作时,H+并不能扩散到二氧化锰的本体深处,只能扩散到表面及其附近,因此MnO2有相当一部分的活性面积无法被利用,使其容量降低.由图7B是不同温度制备的IPC/MnO2复合材料的充放电曲线.从图可知,在60与100°C制备的IPC/MnO2复合材料,其充放电曲线基本保持一致,其比容量也比较接近,分别为405与411 F·g-1.但是30°C制备的IPC/MnO2复合材料比容量相对较低,约为323 F·g-1.合成温度对比容量的影响主要来自于其对材料结构的影响.
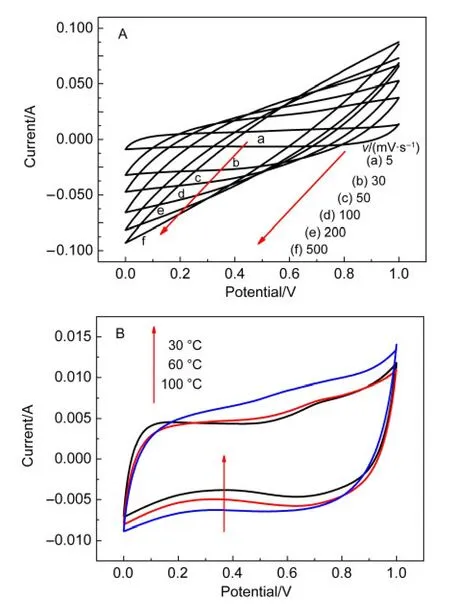
图6 (A)水热温度为100°C时合成的IPC/MnO2复合材料在不同扫描速率(v)下的循环伏安曲线;(B)不同温度下制备的IPC/MnO2复合材料在扫描速率为5 mV·s-1时的循环伏安曲线Fig.6 (A)Cyclic voltammograms of IPC/MnO2at different scanning rates(v)with IPC/MnO2composite prepared at 100°C;(B)typical cyclic voltammograms of the IPC/MnO2composite prepared with the scan rate of 5 mV·s-1at different reaction temperatures
图7 (A)合成温度为100°C时制备的IPC/MnO2复合材料在不同电流密度下的充放电曲线;(B)不同温度制备的IPC/MnO2复合材料在电流密度为5 mA·cm-2时的充放电曲线Fig.7 (A)Charge-discharge curves of IPC/MnO2at different current densities with IPC/MnO2composite prepared at the temperature 100°C;(B)charge-discharge curves of the IPC/MnO2prepared with the current density of 5 mA·cm-2at different reaction temperatures
图8a为IPC/MnO2在温度为30,60及100°C的交流阻抗谱图.由图可见,在高频区,电极阻抗曲线呈半圆状,半径较小;在低频区,交流阻抗曲线是一条大于45°的直线,说明电极反应过程扩散阻抗较小,而且随温度的升高扩散阻抗逐渐减小.图8b表示的是不同反应温度IPC/MnO2复合材料在不同放电电流下的比电容.从图中可以看出,可以看出随着放电电流的增加,比电容有所降低.但是,随着反应温度的提高,100°C的制备温度时在不同的放电电流下其比容量都要优于30和60°C.
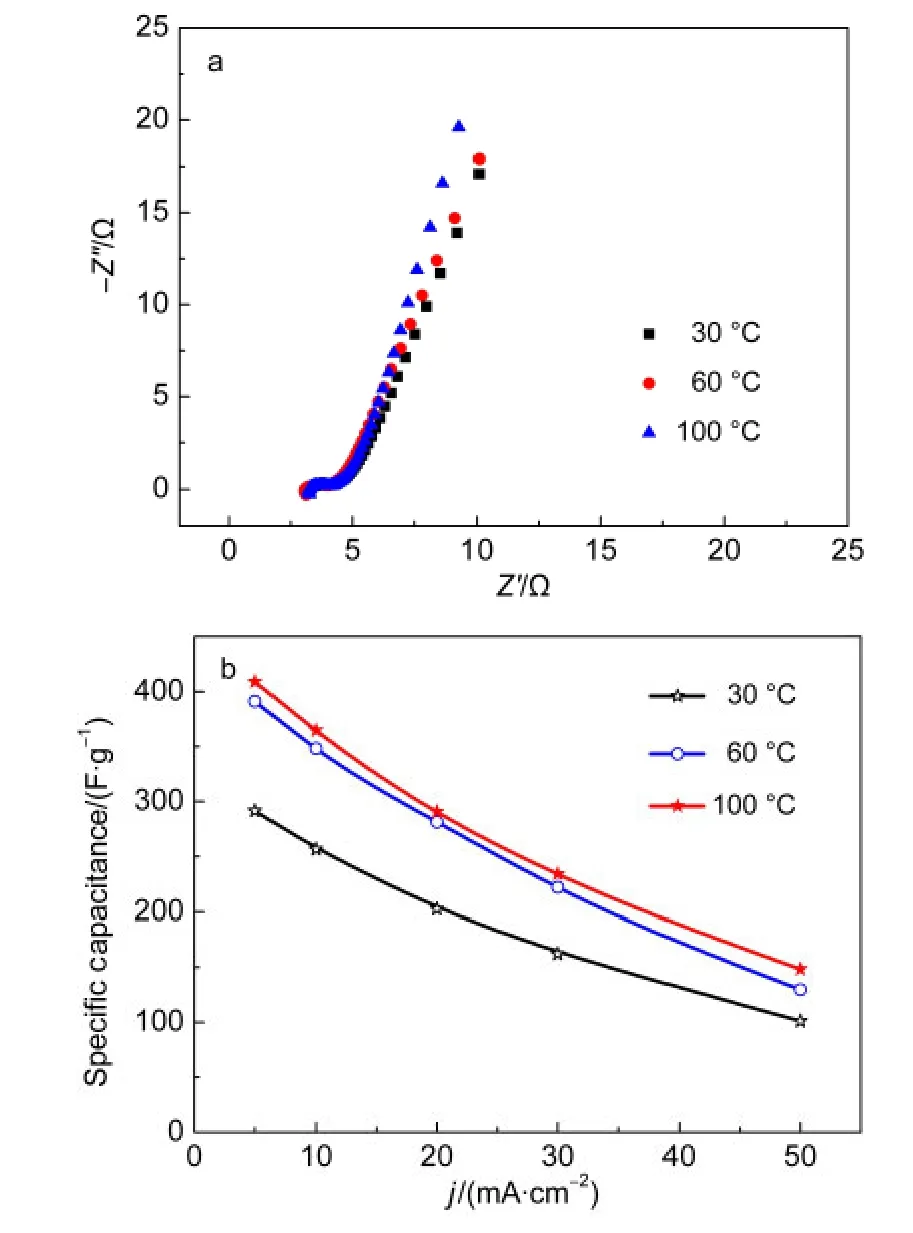
图8 IPC/MnO2复合材料不同温度下的交流阻抗谱(a)和不同放电电流密度(5,10,20,30,50 mA·cm-2)下IPC/MnO2复合材料的比容量曲线(b)Fig.8 EIS(a)and specific capacitance at different current densities of 5,10,20,30,and 50 mA·cm-2(b)of IPC/MnO2 composite prepared at different reaction temperatures
图9为IPC/MnO2复合电极在10 mA·cm-2电流密度下的循环寿命曲线.从图中可以看出在循环500次后,IPC/MnO2复合电极电极的比容量基本保持为原有比容量的60%,表现出了较好的循环寿命.在经过2000次循环后,其比容量基本保持不变,比容量保持为原有比容量的53%.

图9 10 mA·cm-2时IPC/MnO2复合材料的循环寿命曲线Fig.9 Cycle performance of the IPC/MnO2composite at 10 mA·cm-2
3.3 IPC/MnO2//AC混合超级电容器的电化学性能测试
为了进一步评估IPC/MnO2和AC的电化学性能和电位窗口,用循环伏安和恒电流充放电方法分别测试了混合电容器正负单电极的电化学性能.考虑到IPC/MnO2高的比容量及氧化还原特性和活性炭材料快速的离子运输能力,在混合电容器的组装过程中,选择IPC/MnO2复合材料作为正极材料,活性炭(AC)作为负极材料.图10是活性碳电极和IPC/MnO2复合物电极在1 mol·L-1Na2SO4电解液中15 mV·s-1扫描速率下的循环伏安曲线.对比图中曲线,活性碳电极的循环伏安曲线没有氧化还原峰,呈比较规则的矩形,这表明其具有典型的双电层电容特性.IPC/MnO2电极的循环伏安曲线与活性碳电极相似,但是IPC/MnO2呈现的是典型的赝电容特性.由公式(3)计算出IPC/MnO2电极和活性碳电极的放电电荷分别是162和173 F·g-1,则混合电容器的正负极材料质量的理论比为1:0.93;IPC/MnO2电极电极的质量固定为8.0 mg,所以活性碳负极的理论质量约是8.6 mg.另外,在IPC/MnO2电极工作电压范围内没有出现电极极化现象,这说明IPC/MnO2混合氧化物材料适合做宽电位窗口的电容器电极材料.
图11a是IPC/MnO2//AC混合电容器在扫描速率为20 mV·s-1时、不同电位窗口下的循环伏安曲线.从曲线可知混合电容器呈现比较规则的矩形,电位窗口可达1.8 V,为较理想的电容行为.图11b显示的是随着电位窗口的增加,IPC/MnO2//AC混合电容器的比电容的变化情况.根据比电容计算公式(3)可以计算得知,电位窗口从1 V增加到1.8 V,其比电容值从38 F·g-1增加到59 F·g-1,这意味着能量的存储能力及输出功率明显得到改善.另外高的电位窗口为实际应用提供了另一个优势,可以满足一系列电子设备所需的输出电压.34Holze等35也经研究发现AC//MnO2混合电容器在0到1.8 V显示良好的电化学性能.因此,在后续的研究中,我们电位窗口选用1.8 V在1 mol·L-1Na2SO4的电解液中对IPC/MnO2//AC混合电容器进行电化学性能测试.
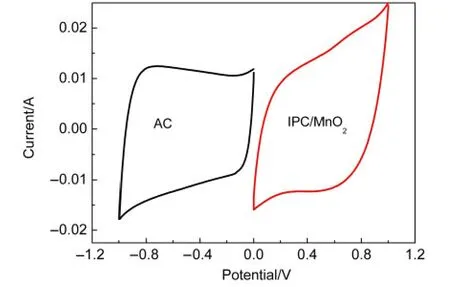
图10 AC和IPC/MnO2电极在扫描速率为15 mV·s-1时的循环伏安曲线Fig.10 CV curves ofAC and IPC/MnO2single electrodes at a scan rate of 15 mV·s-1
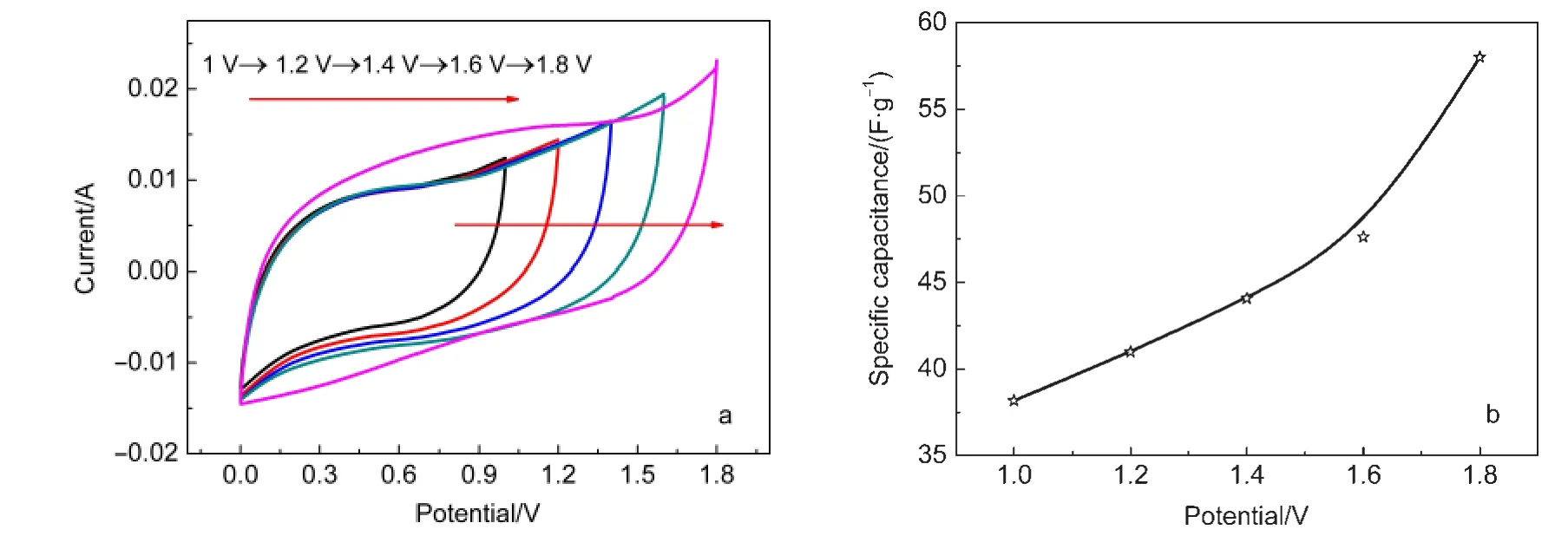
图11 在扫描速率为20 mV·s-1时(a)IPC/MnO2//AC混合超级电容器在不同电位窗口下的循环伏安曲线和(b)混合超级电容器比容量与电位窗口的关系曲线Fig.11 (a)CV curves of an optimized IPC/MnO2//AC asymmetric supercapacitor measured at different potential windows and(b)specific capacitances of the asymmetric supercapacitorwith the increase of potential window at a scan rate of 20 mV·s-1
图12A为IPC/MnO2//AC混合超级电容器在0-1.8 V的电位区间内不同扫描速率(5,10,30,50,100,200,300,500 mV·s-1)下的循环伏安曲线.由图可看出,小扫描速度下的循环伏安曲线基本呈平行四边型,在电位区间内充放电响应电流呈现出了良好的对称性,没有明显的氧化还原峰,这表明在0-1.8 V的电位区间该混合电容器具有典型的电容特性.增大扫描速率后,循环伏安曲线开始变形,充放电电流响应也随扫描速度的增加而增大,标志着快速的电流-电压响应.根据比电容计算公式(3),通过计算可以得到不同扫描速率下的比电容值,图12B显示的是随着扫描速度增加,IPC/MnO2//AC混合电容器的比电容的变化情况.如图所示,随扫描速率的增加,比电容逐渐减少,因为在高的扫描速率下,扩散过程中Na+离子和质子的运动最有可能受时间限制,仅有活性材料的外表面用于电荷存储,导致较低的电化学电活性材料的利用率.扫描速率从5 mV·s-1增加10倍到50 mV·s-1,其比电容值从86 F·g-1减小到39 F·g-1,比电容保持率为45%;但当扫描速率增加100倍到500 mV·s-1时,其比容量只能保持10 F·g-1.值得注意的是在比电容的计算过程中活性物质的质量是两电极材料的总和,而不是单电极的质量.图12C为IPC/MnO2//AC混合超级电容器在不同放电电流密度下的恒流充放电曲线.从图中可以看出,在0-1.8 V充放电压区间,在不同的电流密度下曲线都呈现出良好对称性三角波,这表明其在此区间良好的电容特性和电化学可逆性.但混合电容器的充放电曲线并不像双电层电容那样完美,这是因为存在电极/溶液界面发生的电化学吸脱附和氧化还原反应.由三电极系统测得IPC/MnO2电极的最高比容量为411 F·g-1,但其电位窗口只有1 V,而组装成IPC/MnO2//AC混合超级电容器后,电容器的电位窗口高达1.8 V,这就说明选择合适的正负极材料就可以得到高电位窗口的电容器装置.另外,从图中还可以发现,随着放电电流密度的增加,电容器的电压降增大,比容量也跟着减小.该混合电容器的最大比电容按照公式(2)计算得76 F·g-1,表现出良好的电容特性和大电流放电性能.电化学性能的改善得益于以较大比表面积和适当孔径分布的活性炭为负极,可以促使IPC/MnO2氧化物在较宽的电位窗口内通畅地进行法拉第反应,维持其优异的电容性能.图12D是采用恒电流充放电方法在1 mol·L-1Na2SO4电解液,0-1.8 V的电位窗口内,放电电流密度为10 mA·cm-2下测试的IPC/MnO2//AC混合电容器的循环寿命曲线,从图中可以看出,随着循环次数的增加,混合电容器的比容量逐渐降低.2000次循环后混合电容器的比容量保持为初始比容量的79%,容量仅衰减了21%,显示了IPC/MnO2//AC混合电容器的良好循环稳定性.
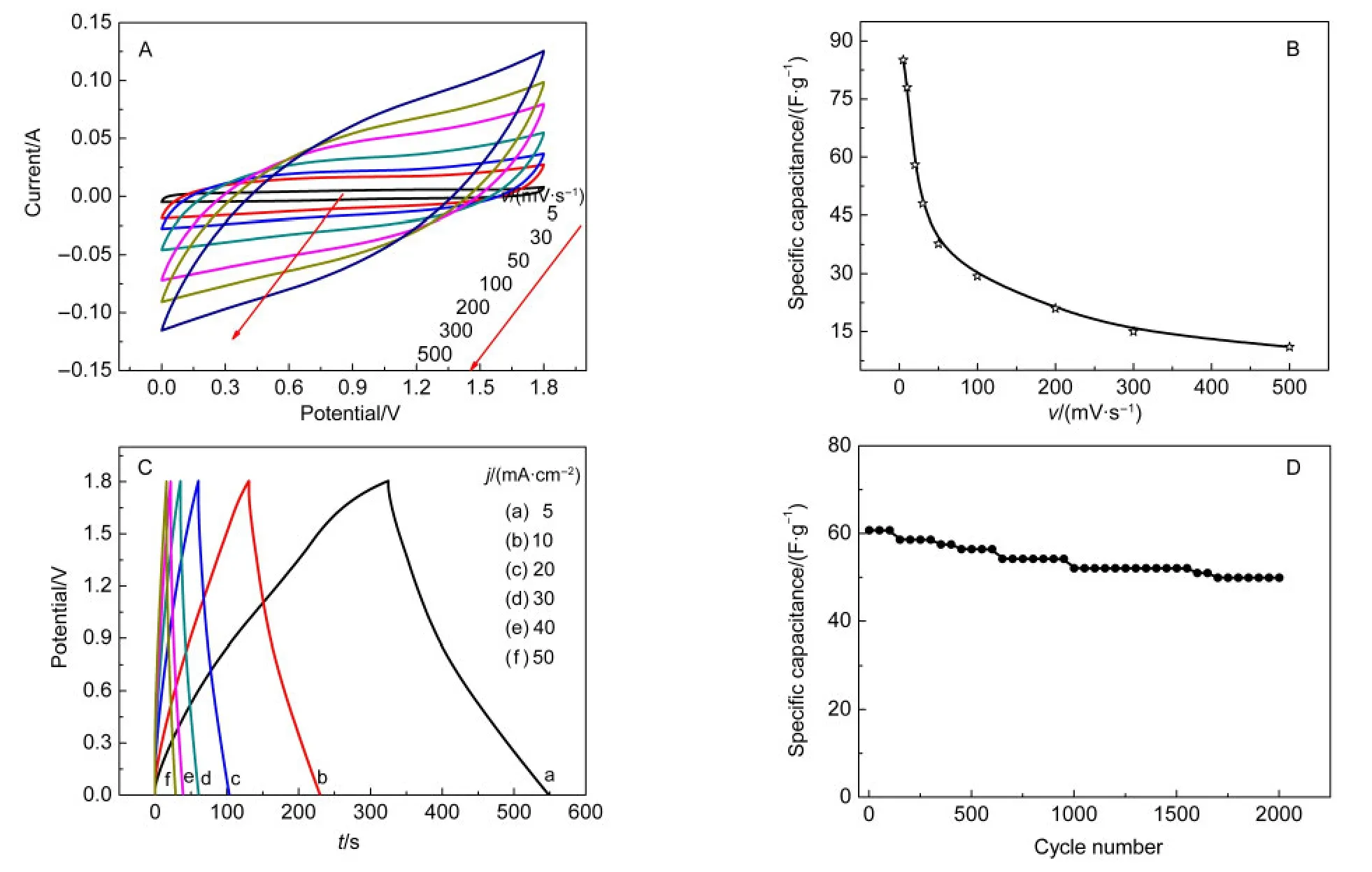
图12 (A)IPC/MnO2//AC混合超级电容器在不同扫描速率下的循环伏安曲线,(B)IPC/MnO2//AC混合超级电容器比容量与扫描速率的关系曲线,(C)IPC/MnO2//AC混合超级电容器在不同电流密度下的充放电曲线,(D)在10 mA·cm-2时IPC/MnO2//AC混合超级电容器的循环寿命曲线Fig.12 (A)CV curves of IPC/MnO2//AC asymmetric supercapacitor measured at different scan rates,(B)comparison of specific capacitances of IPC/MnO2//AC symmetrical supercapacitors at different scan rates,(C)galvanostatic charge/discharge curves of IPC/MnO2//AC asymmetric supercapacitor at different current densities,(D)cycle performance of the IPC/MnO2//AC asymmetric supercapacitorat a current density of 10 mA·cm-2
4 结论
采用水热法合成IPC/MnO2复合电极材料,随着水热反应温度的升高,IPC表面负载的MnO2由纳米颗粒变为卷曲的纳米片状结构.随着反应温度的升高,二氧化锰电极的比容量先增长后基本保持不变;100°C制备的IPC/MnO2复合材料在三电极系统中,电流密度为5 mA·cm-2时,最高比电容达到了411 F·g-1,具有良好的电化学电容性能.组装成IPC/MnO2//AC混合超级电容器,其电位窗口从1 V扩展到1.8 V,容量可达86 F·g-1,且表现出良好的电容特性和大电流放电性能,并具有良好的循环稳定性.2000次循环后,比容量可以保持79%.电化学性能的改善得益于以较大比表面积和适当孔径分布的活性炭为负极,可以促使IPC/MnO2在较宽的电位窗口内通畅地进行法拉第反应,维持其优异的电容性能.
(1) El-Kady,M.F.;Strong,V.;Dubin,S.;Kaner,R.B.Science2012,335,1326.doi:10.1126/science.1216744
(2) Simon,P.;Gogotsi,Y.Nat.Mater.2008,7,845.doi:10.1038/nmat2297
(3)Wen,C.M.;Wen,Z.Y.;You,Z.;Wang,X.F.Chin.J.Chem.Phys.2012,25,209.doi:10.1088/1674-0068/25/02/209-213
(4)Wang,X.F.;You,Z.;Ruan,D.B.Chin.J.Chem.Phys.2005,18,635.
(5) Kim,I.H.;Kim,K.B.J.Electrochem.Soc.2006,153,A383.
(6) Nagarajan,N.;Cheong,M.;Zhitomirsky,I.Mater.Chem.Phys.2007,103(1),47.doi:10.1016/j.matchemphys.2007.01.005
(7)Yu,H.M.;Zheng,W.;Cao,G.S.;Zhao,X.B.Acta Phys.-Chim.Sin.2009,25(11),2186.[余红明,郑 威,曹高劭,赵新兵.物理化学学报,2009,25(11),2186.]doi:10.3866/PKU.WHXB20091113
(8) Fischer,A.E.;Pettigrew,K.A.;Rolison,D.R.;Stroud,R.M.;Long,J.W.Nano Lett.2007,7(2),281.
(9)Sharma,R.K.;Oh,H.S.;Shul,Y.G.;Kim,H.J.Power Sources2007,173,1024.doi:10.1016/j.jpowsour.2007.08.076
(10) Huang,H.J.;Wang,X.Nanoscale2011,3,3185.doi:10.1039/c1nr10229j
(11)Wang,H.L.;Casalongue,H.S.;Liang,Y.Y.;Dai,H.J.J.Am.Chem.Soc.2010,132,7472.doi:10.1021/ja102267j
(12)Sawangphruk,M.;Srimuk,P.;Chiochan,P.;Krittayavathananon,A.;Luanwuthi,S.;Limtrakul,J.Carbon2013,60,109.doi:10.1016/j.carbon.2013.03.062
(13)Yan,J.;Fan,Z.J.;Wei,T.;Qian,W.Z.;Zhang,M.L.;Wei,F.Carbon2010,48,3825.
(14)Song,M.K.;Cheng,S.;Chen,H.Y.;Qin,W.T.;Nam,K.W.;Xu,S.C.;Yang,X.Q.;Bongiorno,A.;Lee,J.;Bai,J.M.;Tyson,T.A.;Cho,J.;Liu,M.L.Nano Lett.2012,12,3483.doi:10.1021/nl300984y
(15) Bordjiba,T.;Belanger,D.J.Electrochem.Soc.2009,156,A378.
(16) Hu,L.B.;Pasta,M.;La Mantia,F.;Cui,L.F.;Jeong,S.;Deshazer,H.D.;Choi,J.W.;Han,S.M.;Cui,Y.Nano Lett.2010,10,708.
(17)Chen,W.;Xie,X.;Liu,N.;Yang,Y.;Wu,H.;Yao,Y.;Pasta,M.;Alshareef,H.N.;Cui,Y.ACS Nano2011,5,8904.doi:10.1021/nn203085j
(18) Prasad,K.R.;Miura,N.J.Power Sources2004,135,354.doi:10.1016/j.jpowsour.2004.04.005
(19)Yan,J.;Fan,Z.;Wei,T.;Qian,W.;Zhang,M.;Wei,F.Carbon2009,47,3371.doi:10.1016/j.carbon.2009.08.001
(20) Yang,Y.J.;Liu,E.H.;Li,L.M.;Huang,Z.Z.;Shen,H.J.;Xiang,X.X.J.Alloy.Compd.2009,487,564.
(21)Yan,J.;Fan,Z.;Wei,T.;Qie,Z.;Wang,S.;Zhang,M.Mater.Sci.Eng.B2008,151,174.doi:10.1016/j.mseb.2008.05.018
(22)Chen,S.;Zhu,J.;Wu,X.;Han,Q.;Wang,X.ACS Nano2010,4,2822.doi:10.1021/nn901311t
(23) Zhang,J.;Jiang,J.;Zhao,X.S.J.Phys.Chem.C2011,115,6448.doi:10.1021/jp200724h
(24) Jin,X.B.;Zhou,W.Z.;Zhang,S.W.;Chen,G.Z.Small2007,3,1513.
(25) Tan,Y.T.;Ran,F.;Kong,L.B.;Liu,J.;Kang,L.Synthetic Metals2012,162,114.doi:10.1016/j.synthmet.2011.11.020
(26)Liu,M.C.;Kong,L.B.;Lu,C.;Li,X.M.;Luo,Y.C.;Kang,L.;Li,X.H.;Walsh,F.C.J.Electrochem.Soc.2012,159,A1.
(27) Li,L.;He,Y.Q.;Chu,X.F.;Li,Y.M.;Sun,F.F.;Huang,H.Z.Acta Phys.-Chim.Sin.2013,29,1681.[李 乐,贺蕴秋,储晓菲,李一鸣,孙芳芳,黄河洲.物理化学学报,2013,29,1681.]doi:10.3866/PKU.WHXB201305223
(28)Khomenko,V.;Raymundo-Pinero,E.;Beguin,F.J.Power Sources2006,153,183.doi:10.1016/j.jpowsour.2005.03.192
(29)Wang,X.;Li,Y.D.Chem.Eur.J.2003,9,300.
(30) Song,X.C.;Zhao,Y.;Zheng,Y.F.Cryst.Growth Des.2007,7,159.doi:10.1021/cg060536h
(31) Ma,R.;Bando,Y.;Zhang,L.;Sasaki,T.Adv.Mater.2004,16,918.
(32)Toupin,M.;Brousse,T.;Bélanger,D.Chem.Mater.2004,16,3184.doi:10.1021/cm049649j
(33) Zolfaghari,A.;Naderi,H.R.;Mortaheb,H.R.J.Electroanal.Chem.2013,697,60.
(34) Izadi-Najafabadi,A.;Yasuda,S.;Kobashi,K.;Yamada,T.;Futaba,D.N.;Hatori,H.;Yumura,M.;Iijima,S.;Hata,K.Adv.Mater.2010,22,E235.
(35)Qu,Q.T.;Zhang,P.;Wang,B.;Chen,Y.H.;Tian,S.;Wu,Y.P.;Holze,R.J.Phys.Chem.C2009,113,14020.
猜你喜欢
食品安全导刊(2021年20期)2021-11-28 00:56:56
新能源汽车供能技术(2021年1期)2021-10-14 08:59:48
物理之友(2020年12期)2020-07-16 05:39:20
电子制作(2019年22期)2020-01-14 03:16:28
山东冶金(2019年5期)2019-11-16 09:09:38
电子制作(2019年23期)2019-02-23 13:21:36
电镀与环保(2016年2期)2017-01-20 08:15:26
现代工业经济和信息化(2016年12期)2016-05-17 05:37:52
通信电源技术(2016年6期)2016-04-20 06:21:10
电源技术(2015年5期)2015-08-22 11:18:02
