
适用于镰刀菌基因敲除和荧光融合蛋白表达的高效通用载体的构建
2014-06-12 03:10:32王晓亮许景升
植物保护 2014年3期
王晓亮, 张 昊, 潘 逸, 许景升, 徐 进, 冯 洁
(中国农业科学院植物保护研究所,植物病虫害生物学国家重点实验室,北京 100193)
丝状真菌是一类产生繁茂菌丝体不产生子实体的真菌,广泛分布于自然界,在工业、农业、医药以及基础生物学研究中具有重要作用[1-2]。禾谷镰刀菌(Fusarium graminearumSchwabe)等植物病原真菌是丝状真菌的重要组成部分,由禾谷镰刀菌引起的小麦赤霉病 (Fusariumhead blight,FHB)是一种世界范围内的重要病害[3],近年来在我国发生尤为严重。该病害的流行除了引起小麦产量损失外,禾谷镰刀菌还能产生多种毒素,造成食品安全上的巨大隐患[4-6]。
禾谷镰刀菌的全基因组测序已于2002年完成,测序菌株为欧洲和北美发现的野生型菌株PH-1,测序结果包括基因组序列、遗传图谱和表达序列标签等[7-9]。在此基础上,采用比较基因组学、生物信息学方法对禾谷镰刀菌生长发育、致病和毒素合成等分子机制的研究成为热点并取得了较大的进展,禾谷镰刀菌等植物病原丝状真菌基因组学的时代已经到来[10]。Wang等系统地对禾谷镰刀菌所有116个蛋白激酶基因进行了功能鉴定,获得了96个基因的突变体,得到了大量表型数据,为进一步明确其参与代谢调控网络奠定了基础[11]。Jiang等鉴定了HOG途径中多个关键基因的功能,明确了其信号转导机制[12-13]。Kimura等阐明了禾谷镰刀菌中单端孢霉烯族毒素合成的分子调控机制[14]。最近,Zhao等利用RNA-Seq技术对于禾谷镰刀菌的基因组的注释进行了修订,修改了655个错误的基因注释,鉴定了2 459个转录活跃区,确定了7 666个基因的5′UTR和3′UTR[15]。与此同时,利用反向遗传学方法,直接从基因入手,通过基因敲除和回复、基因沉默、基因过表达和对相关蛋白进行定位等来研究基因功能的文章也已经十分普遍。王光辉等研究得出小麦赤霉菌中的精氨酸甲基转移酶基因对于赤霉菌菌丝的正常生长和致病性都有重要的作用[16]。Min等研究得出,在禾谷镰刀菌中敲除过氧化物酶体基因PEX5、PEX6和PEX7,影响了碳源营养的利用和致病性[17]。但是,目前对于基因敲除和回复、过表达外源蛋白或荧光融合蛋白,基本上每个基因还需各自独立构建载体,缺少通用的载体,耗费的时间精力非常多[1]。在构建敲除载体的时候,两条同源重组片段需要连接在载体抗性基因的两侧,这样就需要使用4种不同的酶切位点、2次成功的转化来保证重组片段的方向性和正确性(图1)。近些年来,出现了很多依靠不同的限制性内切酶消化出特定酶切位点,再将常规PCR获得同源重组片段插入到这些位点上的高通量的技术,包括了Xi克隆法(Xi-cloning)、连 接 酶 克 隆 法 (LIC-PCR)、重 组 克 隆 法(recombinational cloning)以及USER酶克隆技术(USER friendly cloning techniques)[18-21]。
USER(uracil-specific excision reagent)酶克隆技术,可以在不使用酶切位点和DNA连接酶的条件下直接将PCR产物克隆到载体。USER酶是一种特异性切割尿嘧啶的混合酶,包含尿嘧啶DNA糖基化酶(UDG)和核酸内切酶Ⅷ。其中,UDG催化尿嘧啶碱基的切除,形成一个脱嘧啶位点,但同时保持了磷酸二酯键骨架的完整性。核酸内切酶Ⅷ的裂解酶活性则分别在脱碱基位点的3′端 和5′端裂解磷酸二酯键骨架,释放无碱基的脱氧核糖。此方法需在PCR引物5′端加入一段较长的(8~10bp)包含有脱氧尿嘧啶dU的与载体互补的特异性序列。在USER酶作用下,引物两侧5′端至dU的序列被剪切,形成具有8~10个碱基的3′突出黏性末端,同时在载体上制备与之互补黏性末端,由于黏性末端长度远长于普通酶切位点,所以不需要DNA连接酶的作用即可完成连接。本研究构建了3个适用于USER酶克隆技术的通用载体,并通过试验证明这些载体在不需要酶切位点和DNA连接酶的条件下具有很高的连接效率,可用于大规模构建镰刀菌基因敲除和荧光融合蛋白表达载体,从而进行高通量镰刀菌基因功能分析。

图1 基因敲除载体的两种构建方法Fig.1 Two methods for construction of gene replacement vectors
1 材料与方法
1.1 菌株和DNA
试验使用的小麦赤霉病菌株为禾谷镰刀菌国际标准菌株PH-1。将菌株接种在PDA平板上28℃避光培养5d,刮取菌丝于2mL离心管中,液氮速冻,破碎,采用真菌DNA小量提取试剂盒D3390-02(Omega,美国)提取DNA,-20℃保存备用。
1.2 酶和试剂
USERTM酶,PacI酶,Nt.BbvCI酶(New England Biolabs公司,美国),大肠杆菌(E.coli)感受态细胞DH5α(天根生化有限科技公司,北京),寡核苷酸退火缓冲液(碧云天生物技术研究所),PCR筛选阳性克隆使用PCR MIX(东盛集团有限公司,广州),PRIMERSTAR高保真DNA聚合酶(宝生物公司,大连)所有PCR反应以及温度孵育过程都在Eppendorf Mastercycler gradient(Eppendorf公司,德国)上进行,引物设计根据Broad研究所提供的数据以及相关的注释(网址:http:∥www.broad.mit.edu/annotation)并通过Prime Premier 5进行设计,引物合成和后续的测序都由上海生工生物工程有限公司完成。
1.3 载体构建
pKH质粒和pKN质粒由本实验室保存。pKH质粒是pBluescriptⅡKS质粒为基础,在EcoRⅤ位点插入了潮霉素抗性片段。pKN质粒是pBluescriptⅡKS质粒为基础,在XhoⅠ位点插入G418抗性片段。
1.3.1 基因敲除通用载体的构建
合成互补的寡核苷酸序列UBamHI和UEcoRI、DXhoI和DKpnI,来构建抗性基因两侧的USER酶克隆位点(USER cloning sites,UCS)。100μL退火体系:UBamHI(DXhoI)10μmol/L、UEcoRI(DKpnI)10μmol/L、退火缓冲液1×;退火程序:95℃5min,每8s下降0.1℃,降至25℃,4℃保温,使两条寡核苷酸序列形成双链互补序列,UBamHI和UEcoRI形成USER酶克隆位点1(UCS1),DXhoI和DKpnI形成USER酶克隆位点2(UCS2)。将UCS1连入pKH载体的BamH I和EcoR I位点,UCS2连入XhoI和KpnI位点,转化至大肠杆菌获得单克隆测序验证,构建载体命名为pKH-KO。
1.3.2 荧光融合蛋白表达通用载体的构建
以pGenGFP载体为模版,用PRIMERSTAR高保真DNA聚合酶,GFPFHindⅢ和GFPRKpnⅠ为引物扩增获得大小1 100bp的GFP-Tnos片段,分别连入pKH和pKN两个载体的KpnⅠ和HindⅢ位点之间,转化至大肠杆菌,构建载体分别命名为pKHGFPE和pKNGFPE。
1.4 基因敲除与荧光融合蛋白表达通用载体的应用
1.4.1 通用载体的制备
将含有载体pKH-KO、pKHGFPE和pKNGFPE的大肠杆菌接种于含有氨苄的LB培养基,37℃200r/min过夜培养。采用Axygen质粒小提试剂盒(爱思进生物技术公司,杭州)提取质粒。超微量分光光度计NanoVue(GE公司,美国)检测质粒浓度。采用300μL体系加入70UPacⅠ 后37℃过夜酶切约10μg质粒。第2天,再加入20UPacI和40UNt.BbvCI,37℃温育1h,取2~3μL酶切产物进行电泳检测。然后用Axygen DNA纯化试剂盒进行纯化,得到终浓度约50ng/μL的通用载体,-20℃保存待用。
1.4.2 禾谷镰刀菌FgPex 5基因敲除载体的构建
根据禾谷镰刀菌基因组信息设计FgPex 5基因上游序列扩增引物P5UF、P5UR和下游引物P5DF、P5DR,并在引物的5′端分别添加 O1、O2和O3、O4序列:

以基因组为模版采用东盛taqmix扩增FgPex 5基因的上下游片段P5U和P5D,电泳检测PCR产物浓度。使用25μL体系进行酶切连接,体系包含:

反应条件:37℃温浴20min,25℃温浴20min。反应产物转化大肠杆菌,挑取单克隆测序验证。
1.4.3 禾谷镰刀菌FgPex 5荧光融合表达载体的构建
根据FgPex5基因序列信息设计引物P5PF、P5GR,扩增包含上游启动子与ORF的序列(不含终止子)P5PG,并在引物的5′端分别添加1.4.2中O3和O4序列。
以基因组为模版,采用PfuTurbo®Cx Hot-start DNA Polymerase(Agilent,600410)扩增包含启动子的FgPex 5基因序列P5PG。使用25μL体系进行酶切连接,体系包含:
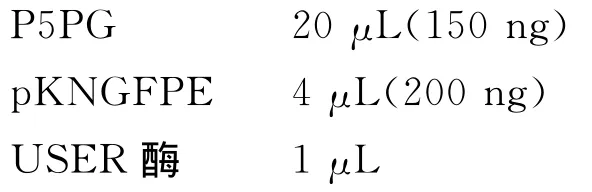
反应条件:37℃温浴20min,25℃温浴20min。反应产物转化大肠杆菌,挑取单克隆测序验证。
1.4.4 原生质体的转化
参考Desmond的方法[22]制备原生质体,并将构建好的载体用KpnⅠ线性化后进行原生质体转化,获得的转化子使用相应的潮霉素B或G418进行抗性筛选。
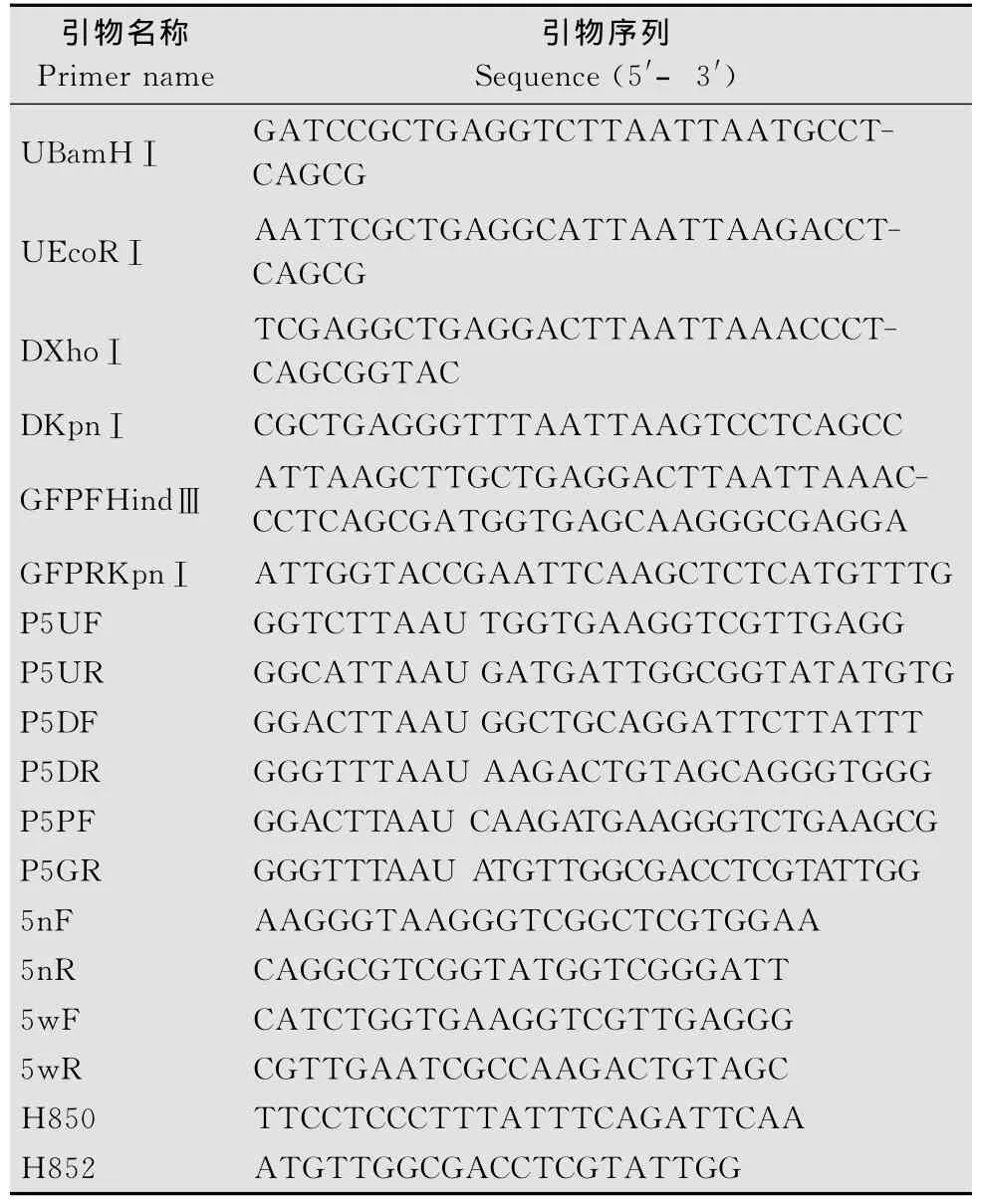
表1 试验所用引物Table 1 Primers for experiments
2 结果与分析
2.1 基因敲除与荧光融合表达载体的构建与应用
构建了基因敲除通用载体pKH-KO(图2a)和荧光融合蛋白表达通用载体pKHGFPE(图2b)和pKNGFPE(图2c)。
本方法通过特定的酶将载体上的USER酶特异位点和两条PCR重组片段产物消化出互补的黏性末端,通过一次连接反应就能将两条同源重组片段连接到抗性基因的两侧。在目的基因的两条同源重组片段扩增引物的5′端加上含有尿嘧啶脱氧核苷酸的9个碱基(图3a),这样PCR获得的同源重组片段在USER酶的作用下就会被消化出特定黏性末端(图3b)。使用PacⅠ和Nt.BbvCI两种酶消化含有UCS序列的通用载体,使其出现与同源重组片段互补的黏性末端(图3c)。由于互补序列长达9个碱基,上述两个消化产物可以在没有DNA连接酶的条件下直接完成连接反应(图3d)。连接产物可以直接进行下一步试验(图3e)。荧光融合表达载体的构建原理与之相同。
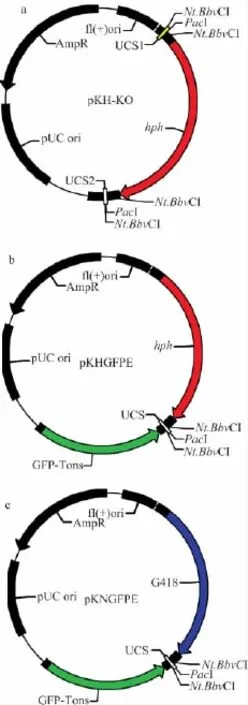
图2 载体的示意图Fig.2 The constructed USER vector

图3 USER酶一步法构建基因敲除载体示意图Fig.3 The USER enzyme strategy for single step construction of gene replacement vectors
2.2 FgPex5基因敲除与GFP融合表达转化子的鉴定
使用基因敲除通用载体pKH-KO在禾谷镰刀菌野生型菌株PH-1中进行FgPex 5基因敲除。对于获得的转化子采用PCR鉴定的方法进行筛选,泳道1、2、3、4为野生型菌株PH-1电泳结果,5、6、7、8为FgPex 5基因缺失突变体菌株电泳结果。用3对引物检测转化子其位置如图4所示,3对引物各自作用如下:
1)引物5nF和5nR是用来检测目的基因是否被敲除(泳道2、6);2)引物H852和H850是用来检测hph基因的存在(泳道1、5);3)引物5wF和H852是用来检测上游的同源重组的发生(泳道3、7);4)引物H850和5wR是用来检测下游的同源重组的发生(泳道4、8)。只有当如图同源重组发生时,目的基因才会被hph基因替换掉。
使用荧光融合蛋白表达通用载体pKNGFPE导入到菌株中,将孢子和萌发的菌丝在荧光共聚焦显微镜(Leica TCS SP2,德国)下观察并拍照,GFP荧光的激发波长在488nm,结果如图5所示。
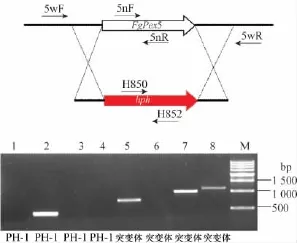
图4 PCR验证检测策略及结果Fig.4 The PCR analysis of mutants

图5 荧光定位蛋白信号图Fig.5Subcellular localization of GFP fusion protein in the mutants
3 结论与讨论
对于真菌基因组测序结果的初步注释已经预测到了大量没有相关同源蛋白的假设蛋白[23]。仅就禾谷镰刀菌PH-1而论,在13 938个假设基因里就有8 091个基因(占58%)是“保守假设蛋白”或者“假设蛋白”一类[24]。面对如此多的假设基因和假设蛋白,我们就需要一个十分高效的基因敲除策略来研究各个基因对于致病性和次级代谢物方面的影响。
采用传统方法构建基因敲除载体,需要至少两次酶切、连接、转化分别连接两个同源重组片段,而本方法将酶切与连接步骤合并,并且仅需一步可以同时连接上两个片段(图1),极大地简化了工作、提高了效率。影响载体构建工作的另一个限制因素是酶切位点的选择,克隆片段中的酶切位点直接影响到载体的构建,一般的载体可以提供多个位点进行选择,但面对全基因组众多基因来说,仍然是远远不够的,特别是荧光融合蛋白表达载体构建中选择酶切位点的同时还要考虑两个基因之间linker序列的碱基数目,这就更加限制了酶切位点的选择。而本方法的另一个优势就在于采用自身构建的9bp互补序列进行连接,完全省去了酶切位点选择的步骤,这为载体的构建从设计上减轻了很大的工作量。同时,相对于普通酶切位点2~3个碱基黏性末端的连接,本方法中长达9bp的黏性末端在不用DNA连接酶的情况下使连接效率得到了很大的提高。近年来split-marker方法[25]构建敲除载体在镰刀菌的基因功能分析中得到了比较广泛的应用[11,26],与传统方法相比,步骤简单,省时省力。但在实际操作过程中发现,不同基因重叠PCR条件差异较大,经常出现重叠失败或者重叠扩增效率低、产物量少,不足以进行下一步转化的情况,摸索最佳条件往往花费很长时间。而本方法所采用的全部是最基础的分子生物学实验技术,成功率高,更易于上手操作。
使用本文的方法进行载体构建由于扩增引物中含有dU,所以需要注意DNA聚合酶的选择。来源于真细菌的Taq酶可以扩增dU,对于片段较短、保真性要求不高的基因敲除载体构建可以选用Taq酶。而构建荧光融合表达载体则需要扩增包含启动子的整个基因,片段较长并且要求不能有碱基错误,这就需要用高保真DNA聚合酶。但一般来源于古细菌的高保真DNA聚合酶如Vent、Pfu等均无法正常扩增dU[27]。因此,本研究选用了目前唯一能够扩增dU的高保真DNA聚合酶PfuTurbo®Cx Hotstart DNA Polymerase(Agilent,600410)进行扩增,取得了很好的效果。
本实验室通过使用USER酶一步载体构建法构建了禾谷镰刀菌十几个过氧化物酶体基因(如Fg-Pex5基因)的敲除载体与绿色荧光蛋白融合表达载体,进行了原生质体的转化,获得了大量的基因敲除转化子,在转化效率上与传统载体构建方法相比没有明显差异。采用这种方法构建目的载体十分适合高通量的基因敲除与荧光融合表达试验[27]。这种方法对于在实验室条件下进行镰刀菌基因敲除和蛋白定位等基因功能的研究有着很大的应用前景。
[1]李海娇,卢建平,刘小红,等.适用于稻瘟病菌基因敲除、过表达和荧光融合蛋白表达载体的构建和使用[J].农业生物技术学报,2012,20(1):94-104.
[2]陈献忠,沈微,樊游,等.后基因组时代的丝状真菌基因组学与代谢工程[J].遗传,2011,33(10):1067-1078.
[3]Watters M K,Randall T A,Margolin B S,et al.Action of repeat-induced point mutation on both strands of a duplex and on tandem duplications of various sizes in Neurospora[J].Genetics,1999,153(2):705-714.
[4]O’Donnell K,Kistler H C,Tacke B K,et al.Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum,the fungus causing wheat scab[J].Proceedings of the National Academy of Sciences of the United States of America,2000,97(14):7905-7910.
[5]Tanaka T,Hasegawa A,Yamamoto S,et al.Worldwide contamination of cereals by the Fusarium mycotoxins nivalenol,deoxynivalenol,and zearalenone.1.Survey of 19countries[J].Journal of Agricultural and Food Chemistry,1988,36(5):979-983.
[6]Windels C E.Economic and social impacts of Fusarium head blight:changing farms and rural communities in the Northern Great Plains[J].Phytopathology,2000,90(1):17-21.
[7]Trail F,Xu J R,Miguel P S,et al.Analysis of expressed sequence tags fromGibberella zeae(anamorph Fusarium graminearum)[J].Fungal Genetics and Biology,2003,38(2):187-197.
[8]Jurgenson J E,Zeller K A,Leslie J F.Expanded genetic map of Gibberella moniliformis (Fusarium verticillioides)[J].Applied and Environmental Microbiology,2002,68(4):1972-1979.
[9]Lee J,Jurgenson J E,Leslie J F,et al.Alignment of genetic and physical maps of Gibberella zeae [J].Applied and Environmental Microbiology,2008,74(8):2349-2359.
[10]Xu J R,Peng Y L,Dickman M B,et al.The dawn of fungal pathogen genomics[J].Annual Review of Phytopathology,2006,44:337-366.
[11]Wang C,Zhang S,Hou R,et al.Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum[J].PLoS Pathogens,2011,7(12):e1002460.
[12]Jiang J,Liu X,Yin Y,et al.Involvement of a velvet protein FgVeA in the regulation of asexual development,lipid and secondary metabolisms and virulence in Fusarium graminearum[J].PloS One,2011,6(11):e28291.
[13]Jiang J H,Yun Y Z,Yang Q Q,et al.A type 2Cprotein phosphatase FgPtc3is involved in cell wall integrity,lipid metabolism,and virulence in Fusarium graminearum[J].PloS One,2011,6(9):e25311.
[14]Kimura M,Tokai T,Takahashi-Ando N,et al.Molecular and genetic studies of Fusariumtrichothecene biosynthesis:pathways,genes,and evolution[J].Bioscience,Biotechnology,and Biochemistry,2007,71(9):2105-2123.
[15]Zhao C,Waalwijk C,de Wit P J,et al.RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum [J].BioMed Central Genomics,2013,14(1):21.
[16]Wang G H,Wang C F,Hou R,et al.The AMT1arginine methyltransferase gene is important for plant infection and normal hyphal growth in Fusarium graminearum [J].PloS One,2012,7(5):e38324.
[17]Min K,Son H,Lee J,et al.Peroxisome function is required for virulence and survival of Fusarium graminearum[J].Molecular Plant-Microbe Interactions,2012,25(12):1617-1627.
[18]Liang X,Teng A,Chen S,et al.Rapid and enzymeless cloning of nucleic acid fragments:United States of America,US 6936470B2[P].2005-08-30.
[19]Aslanidis C,De Jong P J.Ligation-independent cloning of PCR products(LIC-PCR)[J].Nucleic Acids Research,1990,18(20):6069-6074.
[20]Marsischky G,LaBaer J.Many paths to many clones:a comparative look at high-throughput cloning methods[J].Genome Research,2004,14:2020-2028.
[21]Kodumal S J,Patel K G,Reid R,et al.Total synthesis of long DNA sequences:synthesis of a contiguous 32-kb polyketide synthase gene cluster[J].Proceedings of the National Academy of Sciences of the United States of America,2004,101(44):15573-15578.
[22]Desmond O J,Manners J M,Stephens A E,et al.The Fusariummycotoxin deoxynivalenol elicits hydrogen peroxide production,programmed cell death and defence responses in wheat[J].Molecular Plant Pathology,2008,9(4):435-445.
[23]Frandsen R J,Andersson J A,Kristensen M B,et al.Efficient four fragment cloning for the construction of vectors for targeted gene replacement in filamentous fungi[J].BioMed Central Molecular Biology,2008,9:70.
[24]Guldener U,Mannhaupt G,Münsterkütter M,et al.FGDB:a comprehensive fungal genome resource on the plant pathogen Fusarium graminearum[J].Nucleic Acids Research,2006,34:D456-D458.
[25]Catlett N,Lee B N,Yoder O,et al.Split-marker recombination for efficient targeted deletion of fungal genes[J].Fungal Genetics Newsletter,2003,50:9-11.
[26]Son H,Seo Y S,Min K,et al.A phenome-based functional analysis of transcription factors in the cereal head blight fungus,Fusarium graminearum [J].PLoS Pathogens,2011,7(10):e1002310.
[27]Greagg M A,Fogg M J,Panayotou G,et al.A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil[J].Proceedings of the National Academy of Sciences of the United States of America,1999,96(16):9045-9050.
猜你喜欢
辽河(2024年1期)2024-03-04 06:06:13
现代仪器与医疗(2021年2期)2021-07-21 02:19:16
中国当代医药(2020年26期)2020-11-06 07:22:10
生物信息学(2020年2期)2020-07-09 01:27:56
七彩语文·写字与书法(2019年4期)2019-05-24 07:08:01
源流(2018年7期)2018-12-03 13:26:38
青春美文CUTE(2017年2期)2017-11-14 15:59:33
小溪流(画刊)(2017年9期)2017-10-12 18:35:58
分析化学(2015年10期)2015-11-03 07:38:01
鸭绿江(2013年12期)2013-03-11 19:42:10
