
密度泛函理论计算研究丙烯聚合中阻聚剂的作用
2014-06-09 14:25:22付含琦韩晓昱徐人威李化毅朱博超
石油化工 2014年11期
付含琦,韩晓昱,徐人威,李化毅,朱博超
(1.中国石油 兰州化工研究中心,甘肃 兰州 730060;2. 中国科学院 化学研究所 北京分子科学国家实验室 中国科学院工程塑料重点实验室,北京 100190)
密度泛函理论计算研究丙烯聚合中阻聚剂的作用
付含琦1,韩晓昱1,徐人威1,李化毅2,朱博超1
(1.中国石油 兰州化工研究中心,甘肃 兰州 730060;2. 中国科学院 化学研究所 北京分子科学国家实验室 中国科学院工程塑料重点实验室,北京 100190)
以Cl2Ti+C4H9和Cl2Ti+H为活性中心,利用密度泛函理论计算研究了CO,CO2,O2阻聚剂对丙烯聚合的阻聚机理。计算结果表明,丙烯在Cl2Ti+H上的配位和插入反应比在Cl2Ti+C4H9上更容易;CO与两种活性中心的配位能力均很强,CO通过和活性中心配位产生稳定的配合物降低催化剂的聚合活性;CO2在两种活性中心上的配位能力较弱,CO2主要通过在Cl2Ti+C4H9上进行插入反应生成羧基金属化合物达到阻聚作用;O2在活性中心上的插入反应能远大于丙烯的插入反应能,O2插入后通过形成更稳定的金属过氧化物阻碍丙烯的反应。
丙烯聚合;Ziegler-Natta催化剂;密度泛函理论;阻聚剂
目前工业上丙烯聚合主要采用MgCl2负载的TiCl4型催化剂(TiCl4/MgCl2)。根据聚合介质形态的不同,丙烯聚合工艺可分为液相本体法、气相法和浆液法,进一步可细分为Spheripol,Innovene,Unipol,Novelen,Hypol等工艺[1]。丙烯聚合为放热反应,快速撤出反应热是很重要的工程设计内容,虽然现在的工艺中已设计了多种撤热方式,但丙烯聚合时还是不可避免地会发生暴聚,使反应釜温度迅速升高,造成聚丙烯结块堵塞反应器,堵塞严重时甚至造成停车,影响生产的正常进行。为避免暴聚、减小危险,在聚合工艺中一般需设计“杀死系统”,即向反应器内注入毒害催化剂活性的物种,使催化剂快速失活,从而减缓或终止聚合反应。丙烯聚合系统中常用的阻聚剂和丙烯原料中的毒害物质相同(如CO,CO2,O2等)[1]。CO作为阻聚剂常用于研究丙烯聚合的活性中心浓度和聚合动力学[2]。阻聚剂不仅用于丙烯的Ziegler-Natta催化体系中,还用于乙烯的Ziegler-Natta催化体系、烯烃的茂金属催化体系和非茂金属催化剂体系中,即阻聚剂对大多数配位聚合具有广泛适用性。
阻聚剂一般通过与催化剂活性中心作用,阻止丙烯的配位与插入,从而阻止聚合继续进行。TiCl4/MgCl2催化剂催化丙烯聚合的机理一般认为是阳离子Ti+活性中心起主导作用,因此,普通的Lewis碱均对其有强阻聚作用。工业上为了便于在反应釜内注入和去除阻聚剂,常采用气态物质(如CO,CO2,O2)作阻聚剂。其他带活泼质子的气态物质(如NH3和HS等)也可作为阻聚剂,但由于该类物质会与烷基铝反应,且会产生不良气味等,因此工业上一般不采用。CO,CO2,O2在丙烯聚合中的详细作用机理很少被关注。密度泛函理论(DFT)计算在烯烃配位聚合机理研究中被广泛采用[3-13],通过DFT计算可对CO,CO2,O2的阻聚机理做一定的探讨。
本工作以Cl2Ti+C4H9和Cl2Ti+H为活性中心,利用DFT计算研究了CO,CO2,O2阻聚剂对丙烯聚合的阻聚机理。
1 计算方法
所有DFT计算使用Accelrys公司Dmol3程序完成,结构优化和能量计算采用双数字加极化基组,DFT函数选用Becke交换函数和Parr相关函数。能量、最大力和最大位移的收敛值分别为1×10-5Hartree,0.02 Hartree/nm,0.000 5 nm。
TiCl4/MgCl2催化剂中,Ti活性中心和MgCl2晶体通过Cl桥链接。为计算单体在Ti活性中心的立体化学,一般情况下需考虑MgCl2晶体的作用。在MgCl2晶体存在下,Ti活性中心一般表示为Cl4TiR□(□为配位空位,R为聚合物或烷基)[14],Ti为八面体结构,在烷基铝作用下,Ti被还原为三价或二价。计算结果显示,MgCl2载体上的活性中心更可能为Cl3TiR1R2□(R1为聚合物或烷基,R2为硅烷外给电子体)[4],Ti为三价。本工作计算的反应不涉及Ti活性中心的立体化学,为简化计算,略去MgCl2晶体结构,以2个Cl原子为配体,Ti活性中心为四面体。在四面体结构中,参考茂金属催化剂活性中心典型的阳离子活性中心特征[14],将Ti活性中心设计为Cl2Ti+R,Ti中心仍为三价。在TiCl4/MgCl2催化剂中,通常认为活性中心为离子型Ti[14]。另外,由于TiCl4/MgCl2催化剂对Lewis碱性化合物非常敏感,也表明活性中心具有很强的Lewis酸性,即具有阳离子的特性。Cl2Ti+R活性中心不仅对TiCl4/MgCl2催化剂有一定的适用性,对茂金属及后过渡金属催化剂等配位聚合都有一定的适用性。丙烯聚合中的反应见式(1)~(3)。
丙烯聚合中,活性中心Ti最可能的状态有2种:一种是带增长聚合物链的Ti-PP;另一种是与H2链转移反应后形成的Ti-H活性链。在丙烯聚合中,H2为相对分子质量调节剂,H2和Ti活性中心作用,其中,1个H原子转移到增长链上终止链增长,形成聚合物链;1个H原子和Ti链接,形成Ti-H活性中心,继续引发丙烯的插入和聚合反应。
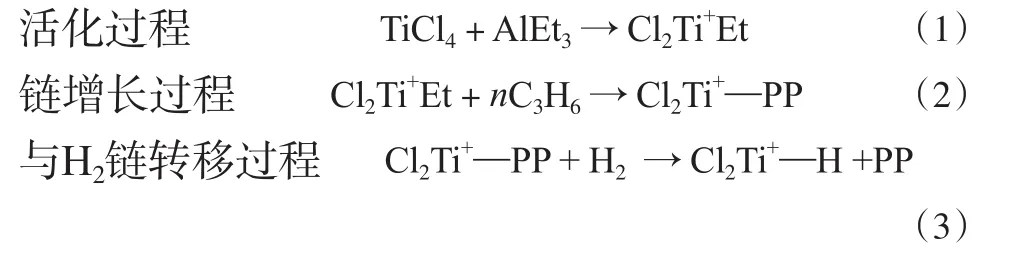
为简化计算,采用异丁基替代聚丙烯链,分别计算2种活性中心(Cl2Ti+C4H9和Cl2Ti+H)和阻聚剂的相互作用。在DFT计算中省略了烷基铝和Ti的相互作用。
2 结果与讨论
2.1 丙烯和活性中心的作用
在丙烯聚合中,活性中心的Ti+处于高度缺电子状态,因此很容易与丙烯中的π电子、阻聚剂的孤对电子作用,丙烯与Ti+作用后,通过进一步插入形成增长链。活性中心与丙烯配位的结构见图1。从图1可看出,在Cl2Ti+C4H9/C3H6中,丙烯双键键长由0.133 9 nm增至0.136 8 nm,说明丙烯配位后被活化,有利于后续的插入反应;Ti—C4H9键长从Cl2Ti+C4H9中的0.202 5 nm减至0.199 6 nm。这主要是因为,在Cl2Ti+C4H9中,Ti+与βH间存在agost作用,使Ti—C4H9键的键长增大,而当Cl2Ti+C4H9与丙烯配位后,agost作用消失,Ti—C4H9键的键长变短。计算结果表明,如没有agost作用,Cl2Ti+C4H9中的Ti—C4H9键的键长应为0.197 9 nm,小于Cl2Ti+C4H9/C3H6中的Ti—C4H9键的键长(0.199 6 nm),说明丙烯可活化Ti—C4H9键。从图1还可看出,Cl2Ti+H与丙烯配位后,Ti—H键的键长从0.166 9 nm增至0.167 8 nm,丙烯双键键长由0.133 9 nm增至0.136 5 nm,说明丙烯和Ti—H键均可被活化,有利于后续的插入反应。
活性中心和不同配体的配位能见表1。从表1可看出,所有配体与Cl2Ti+H的配位能均远大于与Cl2Ti+C4H9的配位能,这主要是因为Cl2Ti+H更加缺电子,结构远不如Cl2Ti+C4H9稳定。

图1 活性中心与丙烯配位的结构(键长单位:nm)Fig.1 Structures of the coordination of active centers with C3H6(bond length: nm).
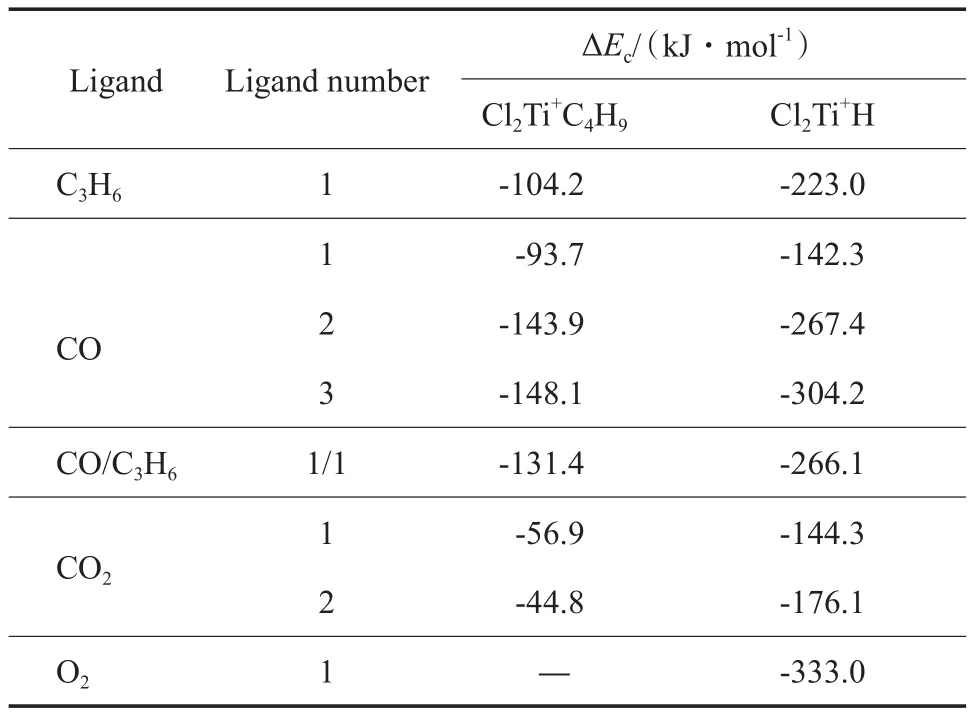
表1 活性中心和不同配体的配位能Table 1 Coordination energies between the active centers and various ligands
活性中心与配体间的插入反应能见表2。从表2可看出,由于Cl2Ti+H的稳定性远低于Cl2Ti+C4H9的稳定性,造成丙烯在Cl2Ti+H上的反应势垒(3.3 kJ/mol)小于在Cl2Ti+C4H9上的反应势垒(17.2 kJ/ mol),且在Cl2Ti+H上的反应能更高(-286.2 kJ/ mol)。结合丙烯在2种活性中心上的配位能可看出,丙烯在Cl2Ti+H上的配位和插入反应均比在Cl2Ti+C4H9上更容易。
2.2 CO和活性中心的作用
DFT计算结果表明,Cl2Ti+C4H9和CO配位时,CO中的C和Ti+的配位能为-93.7 kJ/mol,而O和Ti+的配位能为-34.3 kJ/mol,这说明CO是以C原子而非O原子与Ti+发生作用。活性中心与CO配位的结构见图2。

表2 活性中心与配体间的插入反应能Table 2 Insertion reaction energies between the active centers and ligands

图2 活性中心和CO配位的结构 (键长单位:nm)Fig.2 Structures of the coordination of the active centers with CO(bond length: nm).
从图2可看出,Cl2Ti+C4H9/CO中,Ti—CO,Ti—C4H9,C—O键的键长分别为0.227 9,0.198 5,0.113 5 nm。Cl2Ti+C4H9与CO配位后,Ti—C4H9键长只有很少量的增加,说明CO对Ti—C键没有大的活化作用。CO中C—O键长为0.114 6 nm,在与Ti+C4H9配位后,C—O键长(0.113 2 nm)变短,说明CO上的电子云强烈向Ti+发生转移。
CO的配位能(-93.7 kJ/mol)和丙烯的配位能相当,因此CO可以和丙烯竞争与Cl2Ti+C4H9进行配位。CO与Cl2Ti+C4H9配位后丙烯将很难继续插入,即CO阻断了Cl2Ti+C4H9继续聚合的能力。由于CO分子的体积小,因此可以有1个以上的CO分子与Cl2Ti+C4H9配位。2个CO分子与Cl2Ti+C4H9配位后,Ti—C4H9键长增至0.212 0 nm,配位能为-143.9 kJ/ mol;3个CO分子与Cl2Ti+C4H9配位后,Ti—C4H9键长增至0.217 6 nm,配位能为-148.1 kJ/mol,2个CO分子与Cl2Ti+C4H9配位在能量上更有利。当2个CO分子与Cl2Ti+C4H9配位后,配位能远大于丙烯与Cl2Ti+C4H9的配位能,此时,丙烯就更难以与Cl2Ti+C4H9配位。因此,1~2个CO分子与Cl2Ti+C4H9发生配位都是可能的反应机理,这主要取决于CO的加入量。故在反应中只需加入和Cl2Ti+C4H9数量相当的CO即可使催化剂几乎完全失活,这是CO可高效阻聚的机理。虽然1个CO分子和1个丙烯分子同时在Cl2Ti+C4H9上配位的配位能(-131.4 kJ/mol)和2个CO分子与Cl2Ti+C4H9配位的配位能(-143.9 kJ/ mol)非常接近,但只要CO分子与Cl2Ti+C4H9配位后,分子能量必然大幅降低,使丙烯分子难以继续插入,因此催化活性下降。
在Cl2Ti+H/CO中,Ti—CO,Ti—H,C—O键的键长分别为0.236 3,0.166 5,0.113 2 nm,Ti—H键的键长基本没变,说明CO对Ti—C键没有活化作用;C—O键长变短,说明CO上的电子云强烈向Ti+转移。1个CO分子与Cl2Ti+H的配位能(-142.3 kJ/mol)远小于1个丙烯分子与Cl2Ti+H的配位能(-223.0 kJ/mol)。这主要是因为,丙烯提供了更多的电子云给Cl2Ti+H,对其有较大的稳定作用,而CO提供的电子云较小,但2个CO分子与Cl2Ti+H的配位能(-267.4 kJ/mol)及3个CO分子与Cl2Ti+H的配位能(-304.2 kJ/mol)远大于1个丙烯分子与Cl2Ti+H的配位能。当1个CO与1个丙烯分子同时与Cl2Ti+H配位时,配位能为-266.1 kJ/mol,和2个CO分子与Cl2Ti+H的配位能非常接近。这说明多个CO分子与Cl2Ti+H作用后,将大幅降低丙烯与Cl2Ti+H作用的几率,阻碍丙烯在Cl2Ti+H上的配位和插入反应。
如上所述,当多个CO分子与活性中心配位时,CO占据了Ti原子的空位,且配位能力很强,使丙烯很难再继续配位,丙烯的插入反应被阻断。当1个CO分子和1个丙烯与活性中心配位时,虽然丙烯有继续进行插入反应的可能性,但由于空位被CO占据,丙烯也难以继续插入。因此,CO是通过与活性中心配位产生稳定的配合物,从而阻碍丙烯的插入,降低催化剂的聚合活性。
2.3 CO2与活性中心的作用
活性中心与CO2配位的结构见图3。从图3可看出,Cl2Ti+C4H9与CO2的配位是通过Ti与CO2分子中的O原子作用。Cl2Ti+C4H9/CO2中的Ti—O以及Ti—C4H9键的键长分别为0.223 4,0.204 7 nm;两个C—O 键的键长分别为0.119 1,0.116 2 nm。Cl2Ti+C4H9与CO2的配位能为-56.9 kJ/mol,比其与丙烯和CO的配位能低,这主要是因为CO2分子结构更稳定,给电子能力较弱。Cl2Ti+C4H9/CO2中的Ti—O键的键长比Cl2Ti+C4H9/C3H6中的Ti—C3H6键的键长(见图1)短,主要是因为CO2分子的体积位阻比丙烯更小,更易接近Ti+。CO2分子中C—O键的键长为0.118 0 nm,当CO2与Cl2Ti+C4H9配位后,接近Ti+的C—O键变长,而远离Ti+的C—O键变短,说明CO2上的电子云向Ti+发生转移。Cl2Ti+C4H/CO2中的Ti—C4H9键的键长增大,说明CO2有活化Ti+的作用。如果2个CO2分子与Cl2Ti+C4H9配位,配位能为-44.8 kJ/mol,比1个CO2分子与Cl2Ti+C4H9的配位能低,说明CO2分子更倾向于以单分子形式和Cl2Ti+C4H9作用。CO2与Cl2Ti+C4H9的配位能远低于丙烯、CO与Cl2Ti+C4H9的配位能,因此CO2对丙烯的配位主要为抑制作用,而并非CO那样几乎可阻断丙烯的配位。

图3 活性中心与CO2配位的结构 (键长单位:nm)Fig.3 Structures of the coordinated of the active centers with CO2(bond length: nm).
Cl2Ti+H/CO2中的Ti—O键的键长为0.211 3 nm;Ti—H键的键长为0.165 9 nm;两个C—O键长分别为0.119 9,0.115 5 nm。Cl2Ti+H与CO2的配位能为-144.3 kJ/mol,和Cl2Ti+H与CO的配位能相当;2个CO2分子与Cl2Ti+H的配位能为-176.1 kJ/ mol,小于1个丙烯分子与Cl2Ti+H的配位能。对于Cl2Ti+H,CO2同样只能对丙烯的配位起一定的抑制作用,而并非阻断丙烯的配位。从配位能力推断,CO2对Cl2Ti+C4H9和Cl2Ti+H的阻聚作用均比CO弱。工业试验结果也表明,相比CO,CO2在烯烃聚合阻聚过程中需更多的用量和更长的时间。
活性中心与CO2配位后可通过插入反应生成羧酸金属化合物Cl2Ti+OCOC4H9和Cl2Ti+OCOH,插入反应的过渡态是含Ti+的四元环结构(见图3b和d),反应势垒分别为93.7,39.3 kJ/mol,反应能分别为-163.2,-168.2 kJ /mol(见表2),CO2的插入反应势垒高于丙烯,但反应放热大于势垒,因此CO2的插入反应是放热反应,较容易发生。
可以看出,CO2在活性中心上的配位和插入反应均可与丙烯进行竞争,但在能量上优势并不明显。CO2对丙烯的阻聚作用较弱,是一种较缓和的阻聚剂。对于Cl2Ti+C4H9,CO2的插入反应能(-163.2 kJ /mol)高于丙烯的插入反应能(-106.3 kJ /mol),这必然为丙烯在Cl2Ti+OCOC4H9上的进一步插入带来困难。对于Cl2Ti+H,CO2的插入反应能(-168.2 kJ /mol)小于丙烯的插入反应能(-286.2 kJ /mol),故Cl2Ti+H更易与丙烯反应而不是与CO2反应。结合CO2实际有效的阻聚结果可推测,CO2阻聚作用的主要机理是插入到增长聚丙烯链中形成羧基金属化合物,进而阻碍丙烯的进一步反应。
2.4 O2与活性中心的作用
活性中心与O2配位的结构见图4。从图4可看出,Cl2Ti+C4H9与O2的作用有3种方式:1)O2中的1个O原子与Ti原子形成配合物,Ti—O键的键长为0.202 1 nm,配位能为9.6 kJ /mol,该配合物不稳定。2)2个O原子均与Ti发生作用,O2直接插入到Ti—C键中,形成Cl2Ti+OOC4H9(见图4a),即形成金属过氧键,反应能为-265.7 kJ /mol,为强放热反应。3)1个O原子与Ti活性中心作用,一个O原子与βH作用并发生βH转移反应,形成Cl2Ti+OOHC4H8配合物,即异丁烯分子配位在Cl2Ti+OOH上(见图4b),该反应也没有稳定的配位态和过渡态,反应能为-292.9 kJ /mol。3种方式中,后2种生成过氧键的反应在能量上具有优势,更易发生,且反应能远大于丙烯插入Cl2Ti+C4H9中的反应能。
O2与Cl2Ti+H作用时,可通过单O原子配位,配位能为-27.6 kJ /mol;也可通过双O原子配位,配位能为-333.0 kJ /mol,配位后插入形成Cl2Ti+OOH(图4(c)),反应势垒为4.2 kJ /mol,反应能为-405.8 kJ / mol,反应势垒非常小,说明插入反应易发生。
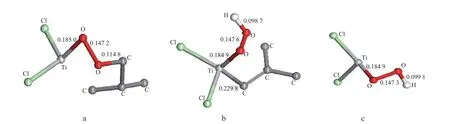
图4 活性中心与O2配位的结构 (键长单位:nm)Fig.4 Structures of the coordination of the active centers with O2(bond length:nm).
O2在活性中心上的插入反应为强放热反应,反应能远高于丙烯的插入反应能,O2插入后形成的金属过氧化物相对更稳定,会阻碍丙烯的进一步反应。Cl2Ti+OOR(R- H或聚合物链)会进一步分解,也可能与烷基铝反应,Cl2Ti+OOH带有活泼氢,很容易与催化体系中的烷基铝继续作用,生成Cl2Ti+OOAlEt2。Cl2Ti+OOR的进一步反应是一个较复杂的体系,需进一步的研究。
3 结论
1)采用异丁基代替聚丙烯链,以Cl2Ti+C4H9和Cl2Ti+H为活性中心,利用DFT计算研究CO,CO2,O2阻聚剂对丙烯聚合的阻聚作用。
2)丙烯在Cl2Ti+H上的配位和插入反应比在Cl2Ti+C4H9上更容易。
3)CO与两种活性中心的配位能力均很强,它通过与活性中心配位产生稳定的配合物从而阻碍丙烯的插入,降低催化剂的聚合活性。CO2在2种活性中心上的配位能力较弱,它主要在Cl2Ti+C4H9上进行插入反应生成羧基金属化合物而达到阻聚作用。O2在活性中心上的插入反应为强放热反应,反应能远高于丙烯的插入反应能,O2插入后形成的金属过氧化物相对更稳定,阻碍丙烯的进一步反应。
[1] 洪定一. 聚丙烯-原理、工艺与技术[M]. 北京:中国石化出版社,2002.
[2] Bukatov G D,Goncharov V S,Zakharov V A. Number of Active-Centers and Propagation Rate Constants in the Propene Polymerization on Supported Ti-Mg Catalysts in the Presence of Hydrogen[J]. Macrom Chem Phys,1995,196(5):1751 -1759.
[3] Zhang Chenggen,Zhang Liaoyun,Li Huayi,et al. Differences Between Insertions of Ethylene into Metallocene and Non-Metallocene Ethylene Polymerization Catalysts[J]. J Phys Organ Chem,2013,26(1):70 - 76.
[4] Wondimagegn T,Ziegler T. The Role of External Alkoxysilane Donors on Stereoselectivity and Molecular Weight in MgCl2-Supported Ziegler-Natta Propylene Polymerization:A Density Functional Theory Study[J]. J Phys Chem C,2012,116(1):1027 - 1033.
[5] 李化毅,张辽云,胡友良. 密度泛函理论研究聚烯烃催化剂取代基电子效应与催化活性的关系[J]. 催化学报,2010(9):1127 - 1131.
[6] Vanka K,Singh G,Iyer D,et al. DFT Study of Lewis Base Interactions with the MgCl2Surface in the Ziegler-Natta Catalytic System:Expanding the Role of the Donors[J]. J PhysChem C,2010,114(37):15771 - 15781.
[7] Li Huayi,Zhang Liaoyun,Wang Zhixiang,et al. Ethylene Polymerization Initiated by Tertiary Diamine/n-Butyllithium Complexes:An Interpretation from Density Functional Theory Study[J]. J Phys Chem A,2010,114(7):2697 - 2700.
[8] Stukalov D V,Zilberberg I L,Zakharov V A. Surface Species of Titanium(Ⅳ) and Titanium(Ⅲ) in MgCl2-Supported Ziegler-Natta Catalysts:A Periodic Density Functional Theory Study[J]. Macromolecules,2009,42(21):8165 - 8171.
[9] Lee J W,Jo W H. Chemical Structure-Stereospecif city Relationship of Internal Donor in Heterogeneous Ziegler-Natta Catalyst for Propylene Polymerization by DFT and MM Calculations[J]. J Organom Chem,2009,694(19):3076 - 3083.
[10] Tomasi S,Razavi A,Ziegler T. Density Functional Theory Investigation into the Stereocontrol of the Syndiospecific Polymerization of Propylene Catalyzed by Cs-Symmetric Zirconocenes[J]. Organometallics,2007,26(8):2024 -2036.
[11] Brambilla L,Zerbi G,Piemontesi F, et al. Structure Of MgCl2-TiCl4Complex in Co-Milled Ziegler-Natta Catalyst Precursors with Different TiCl4Content:Experimental and Theoretical Vibrational Spectra[J]. J Mol Catal,A:Chem,2007,263(1/2):103 - 111.
[12] Bhaduri S,Mukhopadhyay S,Kulkarni S A. Role of Titanium Oxidation States in Polymerization Activity of Ziegler-Natta Catalyst:A Density Functional Study[J]. J Organom Chem,2006,691(12):2810 - 2820.
[13] Flisak Z,Ziegler T. DFT Study of Ethylene and Propylene Copolymerization Over a Heterogeneous Catalyst with a Coordinating Lewis Base[J]. Macromolecules,2005,38(23):9865 - 9872.
[14] Kuran W. Principles of Coordination Polymerisation[B]. Chicthester:Wiley,2011:106 - 113.
(编辑 邓晓音)
Study on the Effects of Inhibitors in Propylene Polymerization by Density Functional Theory
Fu Hanqi1,Han Xiaoyu1,Xu Renwei1,Li Huayi2,Zhu Bochao1
(1. Lanzhou Research Center of PetroChina,Lanzhou Gansu 730060,China 2. Beijing National Laboratory for Molecular Science,CAS Key Laboratory of Engineering Plastics,Institute of Chemistry,Chinese Academy of Science,Beijing 100190,China)
The inhibition mechanisms of inhibitors CO,CO2and O2for propylene polymerization were investigated by means of the density functional theory with Cl2Ti+C4H9and Cl2Ti+H as active centers. The results showed that CO could strongly coordinate with the Ti centers to inhibit the catalyst activity. The coordination ability of CO2with the Ti centers was low, but it could be inserted into Cl2Ti+C4H9and generate Cl2Ti+COOC4H9which was stable and could inhibit the propylene polymerization. O2could react with the Ti centers directly which could generate stable metal peroxides to inhibit the propylene polymerization.
propylene polymerization;Ziegler-Natta catalysts;density functional theory;inhibitor
1000 - 8144(2014)11 - 1259 - 07
TQ 015.9
A
2014 - 05 - 27;[修改稿日期] 2014 - 08 - 14。
付含琦(1981— ),女,四川省资阳市人,硕士,工程师,电话 0931 - 7981983,电邮 31015396@qq.com。联系人:李化毅,电话 010 - 62562697,电邮 lihuayi@ iccas.ac.cn。
猜你喜欢
扬子江诗刊(2023年3期)2023-05-06 10:40:14
大众文艺(2022年16期)2022-09-07 03:08:04
化工设计(2020年5期)2020-11-05 09:41:02
山东化工(2020年8期)2020-06-12 05:19:14
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
农药科学与管理(2019年5期)2019-08-13 00:48:02
石油炼制与化工(2017年5期)2017-04-06 19:47:30
当代化工研究(2016年7期)2016-03-20 16:21:55
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
