
膜萃取与超声衍生联用-负化学源质谱法测定地下水中酚类化合物
2014-06-01 12:30李晓亚桂建业张永涛张辰凌赵国兴田来生
岩矿测试 2014年2期
李晓亚,张 莉,桂建业,张永涛,张辰凌,赵国兴,田来生
(中国地质科学院水文地质环境地质研究所,河北石家庄 050803)
膜萃取与超声衍生联用-负化学源质谱法测定地下水中酚类化合物
李晓亚,张 莉,桂建业,张永涛*,张辰凌,赵国兴,田来生
(中国地质科学院水文地质环境地质研究所,河北石家庄 050803)
酚类化合物由于极性较大,无法直接用气相色谱-质谱法测定,需要通过衍生来降低其极性,提高检测灵敏度;但是传统的衍生步骤繁琐,干扰因素多,操作难度大。本文对传统的前处理方法进行改进,建立了固相膜萃取、超声衍生负化学源质谱测定地下水中酚类化合物的方法。水样萃取后将萃取膜直接放入衍生瓶中,利用超声波的作用力将洗脱和衍生合二为一,超声完成后将溶液直接上机进行测定。测定低、中、高三个浓度水平的加标样品,各目标物的回收率均能达到70%~90%,检出限在0.25~0.35 μg/L之间,相对标准偏差小于10%,能够满足地下水中酚类物质的检测要求。本方法将洗脱和衍生集于一体,使用2 mL丙酮进行洗脱和衍生就可将全部目标物回收,简化了操作步骤,减少了有机溶剂的使用量;同时由于洗脱和衍生是在密闭的环境中进行,外界因素引入的干扰少,克服了二次污染的问题,测定数据更加可靠。
地下水;酚类化合物;固相膜萃取;超声衍生;负化学源质谱法
酚类化合物是芳香烃的羟基衍生物,为原生质毒,对所有生物活性体均能产生毒性,属高毒物质[1],而且它可使水产生异味和颜色,已被许多国家和组织列入环境中优先监测的有机污染物[2-3]。根据国家环境保护部2010年2月发布的《第一次全国污染普查公报》,重点流域(海河、淮河、辽河、太湖、巢湖、滇池)工业污染源的挥发酚排放量高达1938.63万吨,居于各类污染物排放之首[4]。酚类化合物的监测和治理工作任重道远,及时准确测定环境中酚类化合物的含量是预防、控制和治理酚类化合物污染的基础和前提。
酚类化合物的常用测定方法有分光光度法[5-9]、气相色谱法[10-13]、高效液相色谱法[14-15]、气相色谱-质谱法[16-17]等。分光光度法只能测定挥发性酚,不能测定具体的酚类化合物,且操作费时费力。高效液相色谱法在同时分析多种酚类化合物时分离条件比较苛刻,需要通过向流动相中添加缓冲盐并且进行相应的梯度洗脱才能将各种酚类较好地分离。衍生气相色谱-质谱法分离效果好,灵敏度高,但衍生化过程繁琐。本文针对酚类化合物极性大,在GC-MS检测器上响应值低需要衍生的问题,建立了膜萃取与超声衍生联用-负化学源质谱测定地下水中酚类化合物的方法。此方法利用超声波产生的脉动和控制的空化作用增加湍流强度以及相接触面积[18-20],将洗脱与衍生合为一体提高反应速率,大大缩短前处理流程,减少目标物的损失,提高了方法的精密度和准确度。
1 实验部分
1.1 仪器和工作条件
气相色谱-质谱联用仪(GC-MSQP2010,日本岛津公司),配负化学电离源(NCI)。
固相膜萃取装置(德国CNW公司),C18膜(Φ47 mm,美国3M公司),C8膜(Φ47 mm,美国3M公司),SDB-XC膜(Φ47 mm,美国3M公司)。
10 mL刻度衍生瓶,KQ-7100DV数控超声波清洗器(昆山市超声仪器有限公司),水浴锅,MTN -2800W氮吹仪(Auto SCIENCE公司)。
毛细管色谱柱:HP-5MS(30 m×0.25 mm× 0.25 μm)。色谱升温程序:初始温度 90℃,以25℃/min升至300℃,保持1.6 min。进样口温度220℃;进样方式:不分流;进样体积1.0 μL。
质谱条件:离子源类型:负化学源(NCI);反应气:甲烷;质谱扫描方式:选择离子扫描(SIM)。电离能量 70 eV;离子源温度 200℃;连接杆温度220℃。定性定量参数见表1。

表1 定性定量主要参数Table 1 The main qualitative and quantitative parameters
1.2 标准品和主要试剂
标准品:2-甲基苯酚、4-甲基苯酚、2-氯苯酚、4-氯-3-甲基苯酚、2,4-二氯苯酚、2,3,4,6 -四氯苯酚,均购自德国Dr.Ehrenstorfer公司。
二氯甲烷、甲醇、丙酮(色谱纯,德国Merck公司),磷酸(分析纯,石家庄市黄磷厂),五氟苄基溴(99.5%,美国Supelco公司),碳酸钾(分析纯,北京天宇祥瑞科技有限公司),无水硫酸钠(优级纯,天津市巴斯夫化工有限公司)。
1.3 样品前处理
依次用10 mL二氯甲烷、10 mL甲醇和纯水对膜进行活化。在200 mL水样中用50%磷酸调节水样pH在2~4范围,使水样以约200 mL/min速度过膜萃取,萃取后用真空泵将膜充分抽干2 min,然后将萃取膜放入10 mL衍生瓶中,加入2 mL丙酮、30 μL碳酸钾(300 g/L)、100 μL五氟苄基溴(30%,丙酮溶剂),在60℃、功率560 W的条件下超声衍生40 min,然后上机进行测试。
2 结果与讨论
2.1 萃取膜性能分析及萃取回收对比
分别选取适用范围较广的C18膜、C8膜以及适用于极性较强物质的SDB-XC膜进行了萃取性能的比对。水样浓度为0.5 μg/L时(纯水加标样),不同萃取膜的回收率见表2。
C18膜和C8膜的填料为烷基键合固定相,这种填料具有非极性有机配体和极性残余羟基,目标物在固定相上的保留由疏水作用和亲硅醇基作用共同决定,对极性化合物的保留较弱,因此以其为填料的C18、C8膜对多数酚类物质的吸附较差。而SDBXC膜以官能化的、高度交联的、耐受极端pH环境的苯乙烯-二乙烯苯球形聚合物为填料,这是一类高选择性吸附树脂,其衍生化的非极性表面和高交联度会产生较大的比表面积,与C8、C18膜相比,不存在表面残余羟基引起的亲硅醇基次级效应,因此使酚类化合物有较高的回收率。从表2可以看出,使用SDB-XC膜萃取时,各目标物的回收率均能达到70%以上。本实验选择SDB-XC膜。

表2 酚类化合物在不同萃取膜上的回收率Table 2 Recoveries of phenols with different membranes
2.2 pH值对萃取效率的影响
通过pH值的调节可以有效提高吸附剂的选择性和吸附力。相对于硅胶基体吸附剂,改性苯乙烯-二乙烯苯球形聚合物吸附剂,有较强耐受极端pH环境的特性。因此,调节水样pH在2~4范围不会使其性能受到影响,而酚类化合物在此条件下主要以非离子状态存在,回收率能够得以保证,本实验选择萃取pH值为2~4。
2.3 衍生条件
对影响衍生反应的主要因素(时间、超声功率、温度、催化剂等)进行了优化,并通过衍生物的回收率来确定最佳条件。
2.3.1 衍生时间、功率和温度
表3实验结果表明,衍生时间在50 min之内,衍生物的回收率逐渐增大;衍生时间为50 min时,各目标酚类衍生物的回收率均达到最大值。各目标物在50~60℃温度下的衍生效果较好,并且随着超声功率的增大各目标物逐渐显示出较高的回收率。综合考虑,为避免酚的挥发,本实验选择衍生时间为40 min,衍生温度60℃,超声功率560 W。

表3 超声时间、超声功率和衍生温度对回收率的影响Table 3 Effect of ultrasonic time,power and derivization temperature on recovery
2.3.2 催化剂
碳酸钾在衍生反应中起促进催化作用。由于酚类呈现弱酸性,加入碳酸钾这类中性盐会改变反应的环境,这样反应中间体——活化复合物的活度就会有所变化并影响到反应速度,碳酸钾的加入还会影响到弱酸弱碱的电离度从而影响反应速度,这称为中性盐的第一效应和第二效应[21]。实验发现,加入200 μL低浓度碳酸钾(100 g/L)和加入50 μL高浓度碳酸钾(300 g/L),其催化效果后者好于前者(见表4)。这是因为,尽管前者加入的碳酸钾绝对量比后者大,但其引入的水分也较多,水分的存在会导致衍生试剂和衍生产物的水解[22]。因此,在衍生之前充分脱水和尽量少的引入水分是很必要的。本文使用高浓度的碳酸钾(300 g/L),并对其加入量进行试验,确定了催化剂碳酸钾(300 g/L)的加入量为30 μL。

表4 碳酸钾的加入量对回收率的影响Table 4 Effect of K2CO3dosage on recovery
2.4 方法线性范围和检出限
配制浓度范围在10.0~500.0 μg/L之间的酚混合标准溶液,按照1.3节步骤操作,利用外标法对目标化合物的峰面积和浓度作线性回归,分析结果见表5,可以看出各化合物的线性相关系数均大于或等于0.995,线性良好。
配制7个浓度为0.5 μg/L的加标水样进行全流程处理,检出限以3倍标准偏差计算,各目标物的检出限在0.25~0.35 μg/L之间(见表5)。
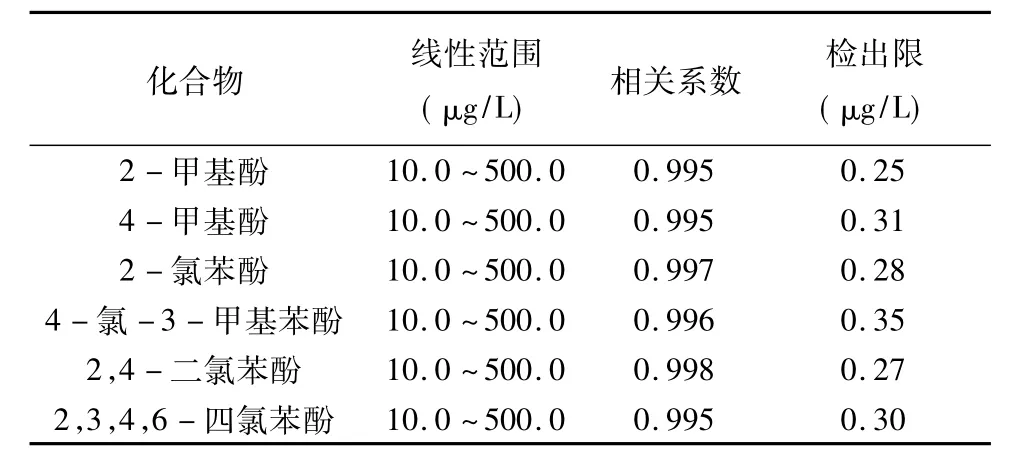
表5 方法线性范围、相关系数、方法检出限Table 5 The linearity range,correlation coefficient and LOD
2.5 方法回收率与精密度
以某厂排污废水(含有2,4-二氯苯酚)200 mL为基质添加低、中、高三个浓度的标准样品溶液,浓度分别为0.1、0.5、2.0 μg/L,按照试验方法全流程处理(浓缩100倍,仪器检测浓度分别为10 μg/L、50 g/L、200 μg/L),测定值减去未加标时的检测结果,除以理论加标量得到回收率,并以7次测定结果计算其精密度。结果见表6,可以看出各目标物在低、中、高三个加标浓度下回收率均能达到70%~90%,相对标准偏差低于10%,能够满足水体中酚类化合物的分析需求。

表6 方法回收率及精密度Table 6 Recovery and precision tests of the method
3 结语
本方法对水中酚类化合物采用固相萃取膜进行萃取,传质速度快,萃取效率高;同时将超声洗脱与衍生联用,使用含有衍生试剂的有机溶剂,借助于超声波的作用力将目标物解吸下来进行衍生反应,本方法各目标物的回收率在70%以上,相对标准偏差在10%以下,能够满足水中酚类物质的检测要求。
本方法将洗脱与衍生步骤合为一体,大大简化了操作步骤,并且前处理实验是在密闭的环境中进行洗脱和衍生,引入的外界干扰因素相对较少,污染少,测定数据更加可靠。与传统的前处理步骤相比具有明显的优势,本方法适用于大批量样品分析。
[1] 高超,王启山,夏海燕.水体中酚类化合物测定方法的研究现状[J].天津化工,2010,24(4):41-42.
[2] 陆蓓蓓,单晓梅,沈登辉,谢继安,张留喜,汤峰.超高效液相色谱-二极管阵列检测器法同时测定水中7种有机酚[J].中国卫生检验杂志,2012,22(6): 1214-1216.
[3] 何淼,饶竹,苏劲,黄毅.GDX-502树脂富集高效液相色谱法测定地表水中酚类化合物[J].岩矿测试,2007,26(2):101-104.
[4] 周艳玲.酚类化合物检测方法研究进展[J].环境监测管理与技术,2011,23(增刊):70-77.
[5] GB/T 5750.4—2006,生活饮用水标准检验方法[S].
[6] Gottlieb S,Marsh B P.Quantitative determination of phenolic fungicides[J].Industrial and Engineering Chemistry,1946,18(1):16-19.
[7] Kidder A G.Determination of yellow color in raw rubber latex films and crepes[J].Analytical Chemistry,1954,26(2):308-311.
[8] Tran D C,Lacerda H D P S.Determination of binding constants of cyclodextrins in room-temperature ionic liquids by near-Infrared spectrometry[J].Analytical Chemistry,2002,74(20):5337-5341.
[9] Simard G R,Hasegawa I,Bandaruk W,Headington E C.Infrared spectrophotometric determination of oil and phenols in water[J].Analytical Chemistry,1951,23(10):1384-1387.
[10] Zhou F R,Li X J,Zeng Z R.Determination of phenolic compounds in wastewater samples using a novel fiber by solidphase microextraction coupled to gaschromatography[J].Analytica Chimica Acta, 2005,538(1-2):63-70.
[11] 李新纪.环境水质中酚类优先监测物的气相色谱法测定[J].色谱,1996,14(1):37-40.
[12] Moder M,Schrader S,Franck U,Popp P.Determination of phenolic compounds in waste water by solid-phase microextraction[J].Journal of Analytical Chemistry,1997,357(3):326-332.
[13] Lee H B,Peart E T,Svoboda L M.Determination of endocrine-disrupting phenols,acidic pharmaceuticals and personal-care products in sewage by solid-phase extraction and gas chromatography-mass spectrometry[J].Journal of Chromatography A,2005,1094(1-2): 122-129.
[14] 苏宇亮,方黎.固相萃取-高效液相色谱法测定水中12种酚类化合物[J].现代科学仪器,2004,96(4): 55-57.
[15] Sun A L,Li J,Liu R M.High-performance liquid chromatographic determination of phenolic compounds in naturalwatercoupled with on-line flow injection membrane extraction-preconcentration[J].Journal of Separation Science,2006,29(7):995-1000.
[16] Saraji M,Bakhshi M.Determination of phenols in water samples by single-drop microextraction followed by insyringe derivatization and gas chromatography-mass spectrometric detection[J].Journal of Chromatography A,2005,1098(1-2):30-37.
[17] Kennison R K,Gibberd R M,Pollnitz P A,Wilkinson L K.Smoke-derived taint in wine:The release of smokederived volatile phenols during fermentation of merlot juice following grapevine exposure to smoke[J].Journal of Agricultural and Food Chemistry,2008,56(16): 7379-7383.
[18] 严伟,李淑芬,田松江.超声波协助提取技术[J].化工进展,2002,21(9):649-651.
[19] US Environmental Protection Agency(EPA).Test Methods for the Analysis of Solid Wastes(SW-864 Method No.3550)[S].1986.
[20] EPA Method 3550B,Ultrasonic Extraction[S].1997:16.
[21] 吴越.催化化学[M].北京:科学出版社,2000.
[22] 谢天民.环境分析化学实验室技术与运营管理[M].北京:中国环境科学出版社,2008.
Determination of Phenols in Groundwater by Solid-Phase Membrane Extraction Combined with Ultrasonic Derivatization-negative Chemical Ionization Mass Spectrometry
LI Xiao-ya,ZHANG Li,GUI Jian-ye,ZHANG Yong-tao*,ZHANG Chen-ling,ZHAO Guo-xing,TIAN Lai-sheng
(Institute of Hydrogeology and Environmental Geology,Chinese Academy of Geological Sciences,Shijiazhuang 050803,China)
Due to their strong polarity,phenolic compounds cannot be directly determined by Gas Chromatography-Mass Spectrometry.Derivatization can reduce the strength of the polarity of phenols,and improve the detection sensitivity.However,the traditional derivative steps are tedious,with many interference factors and complex operations.By applying improvements to the traditional pretreatment method,phenols in groundwater were measured through solid-phase membrane extraction combined with Ultrasonic Derivatization-Negative Chemical Ionization Mass Spectrometry.The membrane was transferred into a derivative bottle after extraction,and then the elution and derivatization were processed simultaneously using ultrasound.The solution was determined by Gas Chromatography-Negative Chemical Ionization Mass Spectrometry directly.By determining the spiked samples at different concentration levels,the recoveries of the method for target components are in the range of 70%-90%,the detection limits are 0.25-0.35 μg/L and relative standard deviation(RSD)is less than 10%.The proposed method simplifies the traditional tedious steps by using 2 mL acetone for elution and derivation.All target objectives have good recoveries.Due to conducting the elution and derivation processes in an airtight environment,less interference factors were introduced without the problem of secondary pollution,and consequently the data were more reliable.
groundwater;phenols;solid-phase membrane extraction;ultrasonic derivatization;Negative Chemical Ionization Mass Spectrometry
P641;O625.31;O657.63
A
0254-5357(2014)02-0270-05
2013-04-01;接受日期:2013-06-24
中国地质科学院水文地质环境地质研究所基本科研业务费项目(SK201204);中国地质调查局项目资助(G201120)
李晓亚,工程师,主要从事地下水有机污染分析。E-mail:lixiaoya0527@126.com。
张永涛,高级工程师,主要从事水质有机分析。E-mail:icpzytws@126.com。
猜你喜欢
河南畜牧兽医(2021年9期)2021-12-10
中国土壤与肥料(2021年5期)2021-12-02
今日农业(2020年22期)2020-12-14
杭州化工(2020年1期)2020-05-09
山东冶金(2019年6期)2020-01-06
中西医结合心血管病杂志(电子版)(2018年25期)2018-01-14
Coco薇(2016年2期)2016-03-22
Coco薇(2015年1期)2015-08-13
小雪花·成长指南(2015年7期)2015-08-11
小雪花·成长指南(2015年4期)2015-05-19
