
中华眼镜蛇神经毒素微球的制备及镇痛作用研究
2014-05-18 08:14:52郝永龙陈美荣
中国药理学通报 2014年5期
郝永龙,陈美荣,黄 铭
(山东中医药大学1.第二附属医院药剂科、2.附属医院眼科,山东 济南 250001)
眼镜蛇神经毒素(α-cobrotoxin,α-CNT)是从中华眼镜蛇毒中(Naja,naja atra)分离出的一种蛋白。该蛋白是由60~70个氨基酸通过4个二硫键连接而成[1]。由于眼镜蛇神经毒素(neurotoxin,NT)特殊的理化性质,使其不易透过血脑屏障(blood-brain barrier,BBB),需要经过一个较长时间的给药过程才能在中枢作用靶位蓄积到较高的药物浓度。在临床中使其应用受到限制。鼻腔给药应用历史悠久[2],鼻黏膜表面积大,黏膜内有丰富的血管,药物利于吸收,无肝脏首过效应,生物利用度高等优点,鼻腔给药除通过体循环透过BBB进入脑组织外,还可通过嗅神经网络通路及嗅黏膜上皮通路而直接吸收入脑,因而可以避开BBB,使常规用药方法难以进入大脑的药物(如水溶性或大分子药物)易于进入大脑,更好地发挥疗效[3]。微球(microsphere,MS)制剂也因其可明显提高药物的生物利用度,近年来在鼻腔给药系统中得到广泛的研究和应用[4-7]。然而,制备微球最常用,包裹效果最好莫过于PLGA(乳酸/羟基乙酸共聚物)。本文主要应用PLGA包裹眼镜蛇毒神经毒素制备微球,通过鼻腔给药,对其镇痛效果进行研究。
1 材料与仪器
1.1 材料 眼镜蛇毒神经毒素(α-CNT):由广西医科大学蛇毒研究所提供。相对分子质量约7 000 u(SDS法),等电聚焦电泳测定 PI为 9.5。PLGA(LA/GA 50∶50,[η]=0.8 dl·g-1)购自德国 Ingelheim公司。海藻酸钠(AGS):AR,25℃下2%溶液黏度为14 000 cps,购自Sigma化学试剂公司。其他化学试剂皆为分析纯。
1.2 仪器 超声波细胞粉碎机:JY92-Ⅱ,上海新芝生物技术研究所。切式匀浆机(高速分散器):XHF-1,上海新芝生物技术研究所。磁力加速搅拌器:78-1型,江苏金坛江南仪器厂。台式高速微型离心机:DGW-99型,宁波新芝科器研究所。冷冻干燥机:LGJ-10,宁波新芝科器研究所。生物型紫外分光光度计:Cary 100 UV-VIS-NIR Spectrophoto-meter,USA。粒径分析仪:Coulter LS-230 Laser,Miami,A-merica。
2 方法
2.1 神经毒素微球制备 采用W/O/O复乳法制备载眼镜蛇神经毒素的PLGA微球,首先称取大约200 mg的PLGA,将其溶解于5 ml乙腈/二氯甲烷(3∶2)的混合溶剂中,得到内油相。然后将1mg眼镜蛇神经毒素溶解于浓度为10 g·L-1海藻酸钠0.5 ml溶液中,得到内水相。将内油相倒入内水相中,经探头式超声混合均匀(100W,5 s×3),得到乳液1。再将该乳液加到10 m l含5%span80的液体石蜡(外油相)中,经匀浆机于2 800 r·min-1下匀浆20 s×2,得到乳液2,然后立刻将乳液2倒入中速搅拌的40 ml含1%span80的液体石蜡中。最终得到的乳液先在室温(18℃左右)下搅拌1 h,然后缓慢升温至35℃,恒温搅拌3~4 h。将固化的微球离心收集,再用石油醚冲洗,以洗去微球表面的液体石蜡。残留的溶剂和石油醚经冷冻干燥机24 h抽干后,于4℃干燥保存。
2.2 药物包埋率的测定 称取50 mg微球,放入1.5 ml含 1%SDS的 NaOH(0.2 mol·L-1)中,充分搅拌后,放入37℃恒温振荡箱中,至微球溶解后取出1 ml溶液,用Lowry法[8]测定眼镜蛇神经毒素吸光度,通过眼镜蛇神经毒素原料药标准曲线来计算微球中所包埋的药物量。
2.3 微球表征研究 ①微球平均粒径及粒径分布测试:将一定量的微球分散于 0.2% (W/V)的Tween 80水溶液中,用粒径分析仪测定粒径。以均数粒径表示其大小。②微球表面形态观察:将干燥后的微球置于试样平台上,喷金后用扫描电镜观察其表面和断面的形态并拍照。
2.4 眼镜蛇神经毒素微球镇痛作用研究 参照文献[9],取大鼠18只,♀♂各半,随机分为3组,分别为α-CNT-MS组、α-CNT组和NS组,分别以α-CNTMS滴鼻剂、α-CNT滴鼻剂及NS,滴鼻70μl,置大鼠于固定筒内,尾部暴露于外,将大鼠尾下部垂直浸入预热的55℃±0.5℃的恒温水浴锅中,浸入长度为4 cm左右,以大鼠甩尾的潜伏期作为痛反应指标。给药前间隔5 min测2次,以其均值为基础痛阈。记录给药后 15、30 min,1、3、6、12、24、48、72、96 h大鼠甩尾潜伏期。
2.5 对鼻黏膜结构变化的研究 大鼠18只,分组给药同上,每日1次,连续给药7 d。d 8每组处死3只动物,取鼻中隔黏膜,观察多日鼻腔给药后局部黏膜结构的损伤程度。d 22处死剩余的动物,取出鼻中隔黏膜组织,观察停药2周后,不用任何保护鼻黏膜的药物,鼻黏膜自然恢复的程度。黏膜取材经处理后进行光学显微镜观察。
3 结果
3.1 微球制备与表征 由于眼镜蛇神经毒素极易溶解于水中,而不溶于乙腈、二氯甲烷等有机溶剂,若采用含水相复乳法(W/O/W),包埋率很低(<20%),药物很快释放进入水相。为了改善眼镜蛇神经毒素的包埋率,必须采用W/O/O非水溶剂复乳法制备。通过应用上述方法我们得到了,眼镜蛇神经毒素微球的包埋率都超过了80%,平均粒径大约在25μm(Tab 1)。通过SEM观察神经毒素微球大小均匀,表面光滑,形态完整,断面致密。如Fig 1。
Tab 1 α-CNT PLGA m icrosphere particle sizeand embedding rate

Tab 1 α-CNT PLGA m icrosphere particle sizeand embedding rate
Embedding rate/% Average particle size/μm 86.4±5.56 25.14±2.64

Fig 1 SEM photographs ofα-cobrotoxin-loaded m icrospheres
3.2 α-CNT-MS对大鼠热刺激痛阈值的影响 用大鼠热刺激甩尾法分别测定α-CNT-MS组、α-CNT组和NS组鼻腔给药前及给药后不同时间的痛阈,见Tab 2。结果显示:α-CNT-MS组和α-CNT组均于鼻腔给药后1 h起效,3 h达峰值,α-CNT组作用持续至12 h,α-CNT-MS组作用持续至72 h,在以上各时间段,大鼠痛阈值增加。与α-CNT组痛阈值相比,差异均有显著性(P<0.05)。经q检验,α-CNTMS鼻腔给药镇痛作用强度明显高于α-CNT,两组3 h时的痛阈值比较P<0.05;其镇痛作用持续时间亦较α-CNT明显延长,24 h、48 h、72 h的痛阈值比较,α-CNT-MS组与NS组和组间的差异均有显著性(P<0.05),而α-CNT组与NS组之间差异无显著性(P>0.05)。
3.3 多日鼻腔给药后对大鼠鼻黏膜结构的影响 3组大鼠分别用 NS、α-CNT、α-CNT-MS鼻腔给药7 d后,肉眼观察鼻黏膜均无红肿、充血等炎症反应,光镜下鼻黏膜组织病理学观察:NS组和α-CNT组均未发现黏膜下及黏膜表面有任何形态学方面的改变;α-CNT-MS组大鼠见鼻黏膜上皮变短、纤毛倒伏,未见炎细胞浸润及上皮细胞坏死脱落;停药2周后光镜观察鼻黏膜无明显异常,纤毛恢复如常,与NS对照组一致。
Tab 2 Influence of nasal drug delivery on thermally stimulated pain threshold of rats
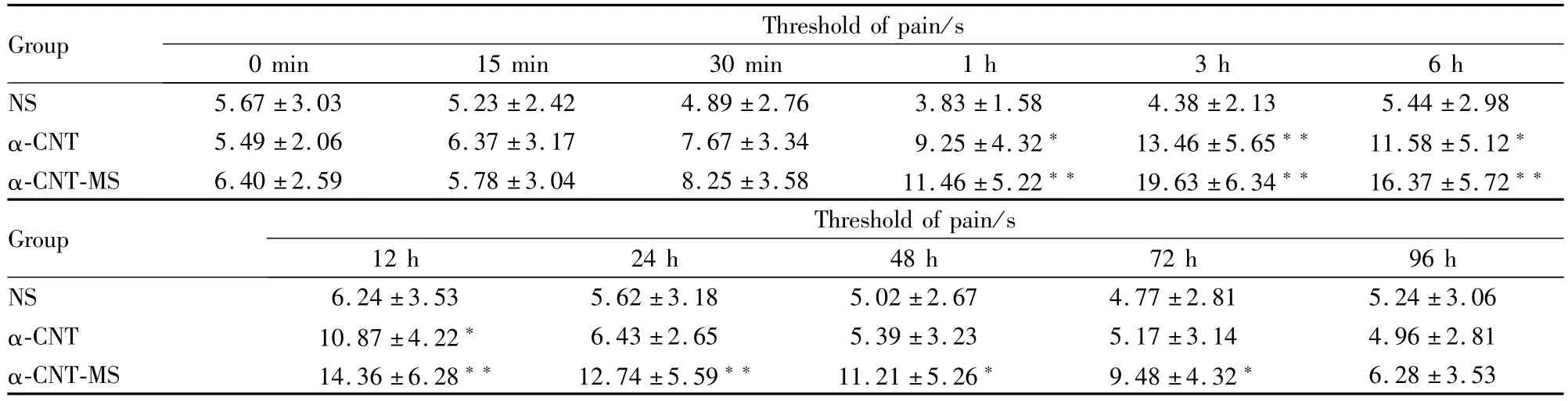
Tab 2 Influence of nasal drug delivery on thermally stimulated pain threshold of rats
*P<0.05,**P<0.01 vs NS group
Group Threshold of pain/s 0 min 15 min 30 min 1 h 3 h 6 h NS 5.67±3.03 5.23±2.42 4.89±2.76 3.83±1.58 4.38±2.13 5.44±2.98 α-CNT 5.49±2.06 6.37±3.17 7.67±3.34 9.25±4.32* 13.46±5.65** 11.58±5.12*α-CNT-MS 6.40±2.59 5.78±3.04 8.25±3.58 11.46±5.22** 19.63±6.34** 16.37±5.72**Group Threshold of pain/s 12 h 24 h 48 h 72 h 96 h NS 6.24±3.53 5.62±3.18 5.02±2.67 4.77±2.81 5.24±3.06 α-CNT 10.87±4.22* 6.43±2.65 5.39±3.23 5.17±3.14 4.96±2.81 α-CNT-MS 14.36±6.28** 12.74±5.59** 11.21±5.26* 9.48±4.32*6.28±3.53
4 讨论
血脑屏障是维持中枢神经系统内环境稳定的结构基础,有效保护脑组织避免外源性有害物质侵害,但也阻碍许多治疗药物进入脑内,限制了中枢神经系统药物的临床应用。如何有效透过血脑屏障成为此类药物发挥治疗作用的关键环节。纳米粒/微球作为一种新型药物载体,能携载药物透过血脑屏障进入脑组织,提高脑内药物浓度,实现脑内靶向给药[10]。PLGA因其良好的生物兼容性、生物可降解性及机械强度,作为制备纳米/微球载体材料引起了人们极大的关注[11]。在微球制剂的研究中PLGA显示出能够控制微球大小、延缓药物降解、延长药物释放时间、靶向释放、降低药物毒性和刺激性等优良特征。本实验中所用的眼镜蛇神经毒素是一小分子单链碱性蛋白,含60~70个氨基酸残基,分子质量7 000 u,等电点约9.5,性质稳定,对热和化学试剂有抗性。为了改善眼镜蛇神经毒素的镇痛效果,提高其生物利用度和减轻其毒副反应,我们选用PLGA为载体材料,采用复乳法制备眼镜蛇神经毒素微球鼻腔给药。由于眼镜蛇神经毒素极易溶解于水中,而不溶于有机溶剂,采用传统的含水相复乳法(W/O/W),药物很快释放进入水相,导致包埋率极低。为此,我们采用了非水溶剂复乳法(W/O/O),并对常规的O/O乳化过程进行了改进,经过反复试验,最终获得了粒径均匀的眼镜蛇神经毒素微球。鼻黏膜作为一种递药系统,因其给药方便,没有首过效应,生物利用度高,受到了国际药学界广泛关注与研究。眼镜蛇神经毒素作为一种大分子蛋白,常规给药途径,难以通过血脑屏障而镇痛效果甚微。本试验就眼镜蛇神经毒素采用PLGA包裹,制备纳/微球,使其黏附时间延长,释放时间延长,通过嗅球及鼻黏膜血管和淋巴管等多条途径,使其透血脑屏障药物增多,达到良好的镇痛效果。本试验药效学结果证实了这一点。
鼻内长期应用药物可能引起鼻黏膜结构、功能的损害,尤其是药物性鼻炎的问题已引起广泛的重视[12],该问题也已成为影响鼻腔给药制剂临床应用的关键问题之一。本试验采用具有良好生物相溶性和生物黏附性的可降解高分子材料PLGA将眼镜蛇神经毒素制成微球制剂,拟利用其较强的黏膜吸附性和缓释特性,使鼻黏膜局部眼镜蛇神经毒素的浓度维持在较低水平,从而降低黏膜毒性。本实验观察了大鼠滴鼻给药1 d,对鼻黏膜结构未产生明显影响;给药7 d后,病理组织切片见鼻黏膜上皮变短、纤毛倒伏,但未见炎细胞浸润及上皮细胞坏死脱落,停药2周后光镜观察鼻黏膜无明显异常,纤毛恢复如常。药物经鼻黏膜吸收可能会在不同程度上对鼻黏膜结构产生损伤,这种损伤是否可逆。如果在给药时注意双侧鼻腔交替使用,避免同一鼻腔长期连续用药,给予其修复再生的时间,可延长鼻腔的用药时间,缩短或减少鼻黏膜受损的时间和程度。
综上所述,应用该方法得到的新型制剂包埋率高,性质稳定,疗效确切,副作用小,可以为大分子蛋白类药物增强疗效,降低毒性提供实验依据,为其在临床应用开发新剂型提供参考。
参考文献:
[1] Yang C C.Cobrotoxin:structure and function[J].J Nat Toxins,1999,8(2):221-33.
[2] Ugwoke M I,Verbeke N,Kinget R.The biopharmaceuticalaspects of nasalmucoadhesive drug delivery[J].Pharm Pharmacol,2001,53:3-21.
[3] 相小强,陶 涛,陈庆华.透血脑屏障制剂的研究进展[J].中国新药杂志,2002,11(7):519-23.
[3] Xiang X Q,Tao T,Chen QH.Rcentadvances of drug delivery systems across blood-brain barrier[J].Chin J New Drugs,2002,11(7):519-23.
[4] 张 奕,蒋新国.鼻腔给药新剂型[J].中国医院药学杂志,1999,19(8):485-8.
[4] Zhang Y,Jiang XG.Drug delivery system for the nasal cavity[J].Chin JHospital Pharm,1999,19(8):485-8.
[5] 李 玲,马海忠,廖明琪,等.鼻腔给药系统类型及临床应用研究进展[J].中国药房,2013,24(17):1615-6.
[5] Li L,Ma H Z,Liao M Q,et al.The nasal drug delivery system as the progress and clinical application[J].China Pharm,2013,24(17):1615-6.
[6] Lim ST,Martin G P,Berry D J,etal.Preparation and evaluation of thein vitrodrug release properties and mucoadhesion of novel microsphere of hyaluronic acid and chitosan[J].Controlled Release,2000,66(2~3):281-92.
[7] Nakanura K,Maitani Y,Lowman A M,er al.Uptake and release of budesonide from mucoadhesive pH-sensitive copolymers and their application to nasal delivery[J].Controlled Release,1999,61(3):329-35.
[8] Lowry O H,Rosenbrough N J,Farr A L,et al.Protein measurementwith folin phenol reagent[J].Biol Chem,1951,193:265.
[9] 徐叔云,卞如濂,陈 修.药理实验方法学[M].第3版.北京:人民卫生出版社,2002:882-904.
[9] Xu SY,Bian R L,Chen X.Methodology of pharmacological experiment[M].3rd edition.BeiJing:People′s medical publishing house,2002:882-904.
[10]王斌艳,夏爱晓,陈苹苹,等.载药纳米粒透过血脑屏障机制的研究进展[J].国际药学研究杂,2010,37(1):40-2.
[10]Wang B Y,Xia A X,Chen PP,etal.Blood-brain barrier transport of drug-loaded nanoparticle:research advances[J].J Int Pharm Res,2010,37(1):40-2.
[11]Leo E,Pecquets S,Rojas J,etal.Changing the pH of the external of the external aqueous phasemaymodulate protein entrapmentand delivery from poly(lactide-co-glycolide)microspheres prepared by a w/o/w solvent evaporation method[J].Microencapsulation,1998,15:421-30.
[12]张 奕,蒋新国.鼻腔给药系统的鼻黏膜毒性及解决途径[J].中国医药工业杂志,2001,32(7):323-7.
[12]Zhang Y,Jiang X G.Detoxification of nasal toxicity of nasal drug delivery system[J].Chin JPharm,2001,32(7):323-7.
猜你喜欢
中老年保健(2021年11期)2021-08-22 03:15:00
潍坊学院学报(2020年6期)2020-11-22 08:04:10
作文大王·低年级(2019年2期)2019-01-23 11:35:18
特别健康(2018年9期)2018-09-26 05:45:40
祝您健康(2018年3期)2018-03-15 22:15:11
作文大王·中高年级(2017年10期)2017-11-21 19:17:09
百科探秘·航空航天(2017年10期)2017-11-08 08:14:29
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国当代医药(2015年29期)2015-03-01 02:07:41
河南科技(2014年22期)2014-02-27 14:18:07
