
系统代谢工程改造树干毕赤酵母生产琥珀酸
2014-05-04 08:05:48王菲菲刘立明
生物加工过程 2014年4期
王菲菲,刘 婷,刘立明
(江南大学 食品科学与技术国家重点实验室 工业生物技术教育部重点实验室食品微生物制造工程研究室,无锡 214122)
琥珀酸作为一种重要的化工原料,广泛应用于食品、制药、农业和化工等领域。同时,还可转化为其他大宗化学品,具有巨大的潜在市场空间[1]。现在,传统的琥珀酸生产存在工艺原料短缺、耗资大和环境污染严重等问题,限制了其进一步发展。近年来,发酵法生产琥珀酸则逐渐成为国内外研究热点,主要集中在产琥珀酸放线杆菌[2]、产琥珀酸曼氏杆菌[3-4]、产琥珀酸厌氧螺菌[5]和重组大肠杆菌[6-7]等。然而,由于这些微生物存在发酵、纯化工艺复杂等原因,限制了其在工业化生产中的应用[8-10]。树干毕赤酵母(Pichia stipitis)是能利用木糖的优势菌株,而木糖广泛存在于林业及农业废弃物中,因此,研究代谢改造树干毕赤酵母生产琥珀酸对生物质资源的利用具有重要意义。此外,P.stipitis具有耐受低 pH、高糖等优点[11],能极大地简化下游工序、降低生产成本,是发酵生产琥珀酸的潜在优势菌株。
本研究中笔者借助树干毕赤酵母全基因组代谢网络模型iTL885[12],设计琥珀酸最佳合成途径,进而利用代谢工程策略对树干毕赤酵母进行代谢改造,以期提高琥珀酸产量。
1 材料与方法
1.1 菌种和质粒
P.stipitis FPL-UC7[13],由美国威斯康辛大学Jeffries教授惠赠;E.coli JM109,质粒 pY26TEFGPD[14]、pUG72由笔者所在食品微生物制造工程实验室保存。
1.2 主要试剂和仪器
PCR仪(Bio-Rad公司),高效液相色谱仪(DIONEX UItimate 3000),5 L发酵罐(上海宝兴生物设备工程有限公司)。
所用内切酶、PCR酶及胶回收试剂盒,大连宝生物公司(TaKaRa);酵母基因组提取试剂盒和质粒提取试剂盒,北京天根生化科技有限公司;琥珀酸脱氢酶试剂盒,南京建成科技有限公司;胰蛋白胨和酵母粉,英国OXOID公司;其他生化试剂,上海生工生物工程有限公司;测序工作由上海华大基因有限公司完成。
1.3 代谢网络模型模拟与分析
模型iTL885[14]是笔者所在实验室成员根据P.stipitis CBS6054基因组序列构建的,包含885个基因、870个代谢物和1 240个代谢反应。
采取iTL885模拟分析高产琥珀酸代谢工程策略,利用COBRA Toolbox将模型iTL885 Excel文件读入Matlab中转化为系数矩阵,并以流量平衡分析方法(FBA)[15]进行模拟分析,约束条件下求解目标方程,获得代谢反应流量分布。模拟的约束条件来源于实验数据,模拟基因过量表达时,将基因对应的生化反应代谢流量设置为原流量的5倍,模拟基因敲除时,将基因控制的生化反应的代谢流量设为0。
1.4 重组菌株的构建方法
1.4.1 重组菌株FPLicl的构建
参照P.stipitis CBS6054 icl1基因序列,设计带有BamH I和EcoR I酶切位点的引物ICL-F/ICL-R(表1),以 P.stipitis FPL-UC7基因组为模板,利用PCR获得目的基因片段。将纯化后的icl1基因片段和载体质粒pY26TEF-GPD进行BamH I和EcoR I双酶切,柱式纯化回收,16℃连接过夜,将连接产物转化到JM109感受态细胞中后,涂布带有氨苄青霉素的LB平板,挑取转化子接种到LB液体培养基(含氨苄)摇瓶中培养,提取质粒酶切验证,测序验证,从而获得表达质粒pY26-icl1。
采用电转化法将构建的重组质粒转化到P.stipitis FPL-UC7感受态中,电转条件:电压1.5 kV,电容25 μF,电阻200 Ω,电击4 ms。经过SC培养基筛选及质粒提取酶切验证,得到重组菌株FPLicl1。
1.4.2 重组菌株FPLΔsdh的构建
采用同源重组的方法敲除出发菌株FPL-UC7的琥珀酸脱氢酶基因(sdh1),由于P.stipitis的同源重组率比较低,因此要扩增相对较大的同源臂与尿嘧啶标记基因融合,构成敲除组件。具体步骤如下:参照P.stipitis CBS6054基因组序列,分别设计扩增琥珀酸脱氢酶基因(sdh1)的上下游800 bp DNA片段的引物,引物分别为upper-F/upper-R、lower-F/lower-R,参照质粒pUG72序列设计扩增尿嘧啶基因片段的引物,引物为ura-F/ura-R,分别PCR得到3个目的片段,质量分数为1%的琼脂糖凝胶电泳回收,以融合PCR方法将3个片段融合连接起来,构成敲除组件,琼脂糖凝胶回收并测序。
将敲除片段电转化P.stipitis FPL-UC7感受态,经过筛选获得转化子,再分别用两对验证引物YF1/Y-R1和Y-F2/Y-R2进行验证,从而获得正确的重组菌株FPLΔsdh。
1.4.3 重组菌株FPLΔsdh-icl的构建
同样采取同源重组的方法使重组菌株FPLΔsdh重新获得尿嘧啶标记。设计具有同源臂的引物upper-F/upper-A和lower-S/lower-R,分别扩增sdh1的上下游序列800 bp,得到的目的片段凝胶回收,融合PCR法进行拼接,构成尿嘧啶标记敲除组件,琼脂糖凝胶回收并测序。将构建好的敲除片段电转化到P.stipitis FPLΔsdh感受态中,转化液经后培养后,涂布5-氟乳清酸(5-FOA)筛选平板,30℃培养,挑取其中较大的转化子传代培养,筛选得到的转化子摇瓶培养提取基因组,进行PCR验证,最后得到恢复尿嘧啶缺失标记的重组菌株FPLΔsdhΔura。
接下来将重组质粒pY26-icl1电转化到FPLΔsdhΔura感受态中,用 SC筛选培养基传代培养,提取质粒验证,最终得到sdh1基因缺失同时过量表达icl1基因的重组菌株PPLΔsdh-icl。
1.4.4 对照菌FPLpy26的构建
将空质粒pY26TEF-GPD转化到P.stipitis FPLUC7感受态中,平板筛选转化子并进行传代培养,得到表达ura3基因能在基本培养基上生长的对照菌FPLpy26。
本实验所使用的引物见表1。

表1 本实验所用到的引物Table 1 Primers used in the experiment
1.5 培养条件
1.5.1 培养基
E.coli培养与保存培养基为LB培养基(g/L):酵母膏5、蛋白胨10、NaCl 10。加入50 mg/L氨苄青霉素的LB用于筛选转化子。
P.stipitis转化子的筛选(SC)培养基(g/L):木糖20、YNB(不含 (NH4)2SO4)1.7、(NH4)2SO45。
5-FOA筛选培养基:SC培养基加入50 mg/L尿嘧啶和1 g/L 5-氟乳清酸;5-FOA用二甲基亚砜溶解,过滤除菌。
P.stipitis种子培养基(YPX)(g/L):酵母粉10、胰蛋白胨20、木糖20。
P.stipitis发酵培养基(YNBX)(g/L):YNB 1.7、尿素 2.27、木糖50。
1.5.2 培养方法
E.coli培养方法:挑取平板活化的单菌落接种到装有25 mL LB培养基的250 mL摇瓶中,37℃、200 r/min培养,培养携带质粒的菌种时,培养基中要加氨苄青霉素。
P.stipitis发酵培养方法:挑取平板活化的单菌落到种子培养基,于30℃、200 r/min摇瓶培养24 h,以体积分数5%接种量接种到5 L发酵罐发酵培养,温度为30℃,pH为6.5,转速为400 r/min,通气量为1.0 vvm(vvm表示每分钟通气量与罐体实际料液体积的比值)。
1.6 分析方法
1.6.1 酶活的测定方法
粗酶液制备:取对数生长中期3 mL发酵液于离心管中,8 000 r/min、5 min离心,去除上清液,菌体用PBS缓冲液(pH 7.4)重悬洗涤并离心2次,去除上清,菌体加入2 mL缓冲液,超声破碎20 min,4 000 r/min离心8 min,上清即粗酶液。总蛋白浓度测定参照考马斯亮蓝法,以牛血清蛋白(BSA)做蛋白浓度标准曲线。
异柠檬酸裂解酶酶活测定参照文献[16]。酶活定义:1 min内氧化1 μmol NADH所需的酶量定义为1 U。比酶活定义为每毫克蛋白含有的酶活(U/mg)。
琥珀酸脱氢酶酶活测定参照琥珀酸脱氢酶试剂盒测定,测定粗酶液于600 nm吸光度的变化值。酶活定义:每毫克蛋白每分钟使反应体系的吸光度降低0.1为1个比酶活单位。
1.6.2 发酵及代谢物分析
细胞生长用分光光度计于600 nm处测定吸光值,细胞干质量(DCW)换算公式为:ρ(DCW)=OD600×0.21。发酵液中相关代谢物质的浓度用液相检测。检测条件:色谱柱为 Aminex HPX-87H(Bio-Rad公司),流动相 0.005 mol/L H2SO4,流速0.6 mL/min,柱温35℃,检测器紫外检测器、示差折光检测器,紫外检测波长210 nm。琥珀酸检测用紫外检测器,乙酸、木糖检测用示差折光检测器。
2 结果与讨论
2.1 基于GSMM模型 iTL885的琥珀酸最佳合成途径的设计
将对照菌FPLpy26于恒化培养器中进行培养,测定培养过程中细胞浓度及木糖浓度随发酵时间变化规律。最大比生长速率(0.10 h-1)时,木糖比消耗速率为2.23 mmol/(g·h)。将木糖消耗速率和琥珀酸合成速率作为模型约束条件,以生物量方程为目标方程,通过FBA计算获得模型iTL885中不同琥珀酸合成途径中代谢流量的分布,如图1(a)所示,琥珀酸比合成速率为 8.9×10-3mmol/(g·h)。进一步借助FBA模拟关键节点处代谢改造对碳代谢流分布及琥珀酸合成的影响,结果发现:①过量表达异柠檬酸裂解酶基因icl1增加乙醛酸(TCA)循环代谢流量,琥珀酸合成速率从 8.9×10-3mmol/(g·h)增大到 0.02 mmol/(g·h)(图 1(b));②敲除sdh1基因能阻断三羧酸循环中琥珀酸的进一步代谢,增大三羧酸循环-乙醛酸循环流量,琥珀酸合成速增加到0.15 mmol/(g·h)(图1(c));③在过量表达icl1基因的同时敲除sdh1基因,可使琥珀酸合成速率提高到0.19 mmol/(g·h)(图1(d))。
2.2 构建高效合成琥珀酸的树干毕赤酵母重组菌株
根据2.1模拟结果,提高树干毕赤酵母生产琥珀酸产量的代谢工程策略包括:①过量表达异柠檬酸裂解酶基因;②敲除琥珀酸脱氢酶基因;③同时过量表达异柠檬酸裂解酶基因和敲除琥珀酸脱氢酶基因。本研究借助代谢工程策略构建了树干毕赤酵母重组菌株 FPLicl、FPLΔsdh 和 FPLΔsdh-icl。
2.2.1 构建过量表达icl1的重组菌株FPLicl
以P.stipitis FPL-UC7基因组为模板,按照方法1.4.1进行PCR,扩增产物进行琼脂糖凝胶电泳,PCR和酶切电泳结果如图2所示。由图2可知:扩增产物大小约1 700 bp,经测序鉴定为P.stipitis icl1基因,无点突变。将扩增片段和载体 pY26TEF-GPD进行BamH I和EcoR I双酶切,经纯化后连接、转化。提取转化子质粒进行酶切验证,得到约7 430和1 700 bp大小的片段,分别为载体和基因片段,结果表明表达载体pY26-icl1构建成功。将重组质粒pY26-icl1转化到P.stipitisFPL-UC7中构建,获得过量表达异柠檬酸裂解酶基因的重组菌株FPLicl。
2.2.2 构建缺失sdh1基因的重组菌株FPLΔsdh
按照方法1.4.2敲除菌株FPL-UC7的sdh1基因,敲除步骤如图3(a)所示。以FPL-UC7基因组为模板,分别扩增得到琥珀酸脱氢酶(sdh1)的上下游800 bp的片段(加引物同源臂为825 bp),以pUG72为模板扩增出尿嘧啶基因片段1 050 bp(图3(b)),测序结果与NCBI上对应序列100%匹配。凝胶回收3段片段,通过融合PCR将3个片段连接起来,构成敲除框,大小为2 600 bp(图3(c))。将敲除框DNA片段转化到P.stipitisFPL-UC7中,通过引物验证转化子基因敲除的正确性,构建获得重组菌株 FPLΔsdh。
2.2.3 构建重组菌株FPLΔsdh-icl
首先对重组菌株FPLΔsdh进行分子操作,按照1.4.3的方法敲除尿嘧啶基因,构建琥珀酸脱氢酶和尿嘧啶双重缺陷型重组菌株FPLΔsdhΔura。提取FPLΔsdh和转化子基因组,用引物upper-F/lower-R进行PCR验证,出发菌扩增出2 600 bp的DNA片段,重组菌株扩增出1 600 bp的DNA片段,通过测序证明重组菌株FPLΔsdhΔura构建完成,图4为尿嘧啶敲除示意图和PCR验证电泳图。然后将重组质粒pY26-icl1转化到 FPLΔsdhΔura感受态中,用SC筛选培养基传代培养,提取质粒验证,获得重组菌株 FPLΔsdh-icl。
2.3 重组菌株酶活测定
于30℃、200 r/min条件下将菌株培养到对数生长中期,取样测定出发菌株和重组菌株中异柠檬酸裂解酶和琥珀酸脱氢酶的活性,结果列于表2。由表2可知:①重组菌株FPLicl中异柠檬酸裂解酶活性为(1.6±0.20)U/mg,是出发菌株的4.8倍,而琥珀酸脱氢酶活性则比出发菌株降低了22%,表明过量表达icl1基因将减弱琥珀酸到富马酸代谢途径;②重组菌株FPLΔsdh琥珀酸脱氢酶活性较低,几乎无法检测;而异柠檬酸裂解酶酶活为(5.6±0.13)U/mg,是出发菌株的17倍;③重组菌株FPLΔsdh-icl的异柠檬酸裂解酶酶活为(6.6±0.17)U/mg,是出发菌株的20倍。
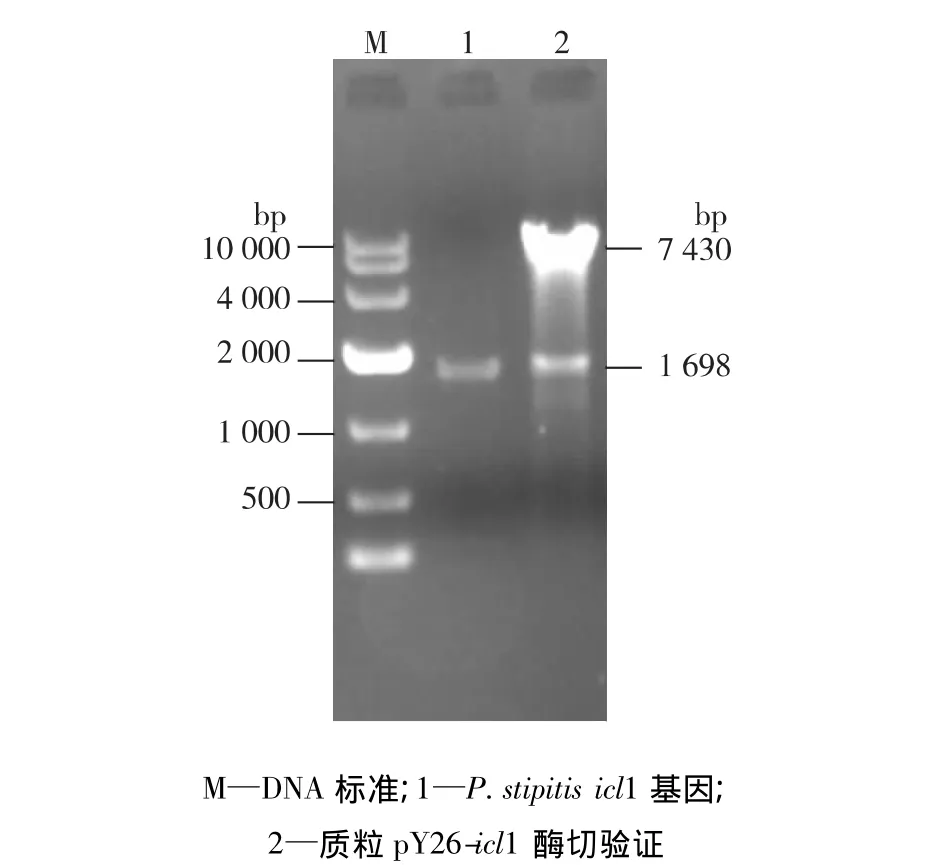
图2 P.stipitis icl1基因PCR扩增和质粒pY26-icl1双酶切验证Fig.2 PCR amplification of icl1 gene and identification of pY26-icl1 by restriction endonuclease digestion

图3 P.stipitis sdh1基因的敲除Fig.3 Deletion of sdh1 gene in P.stipitis
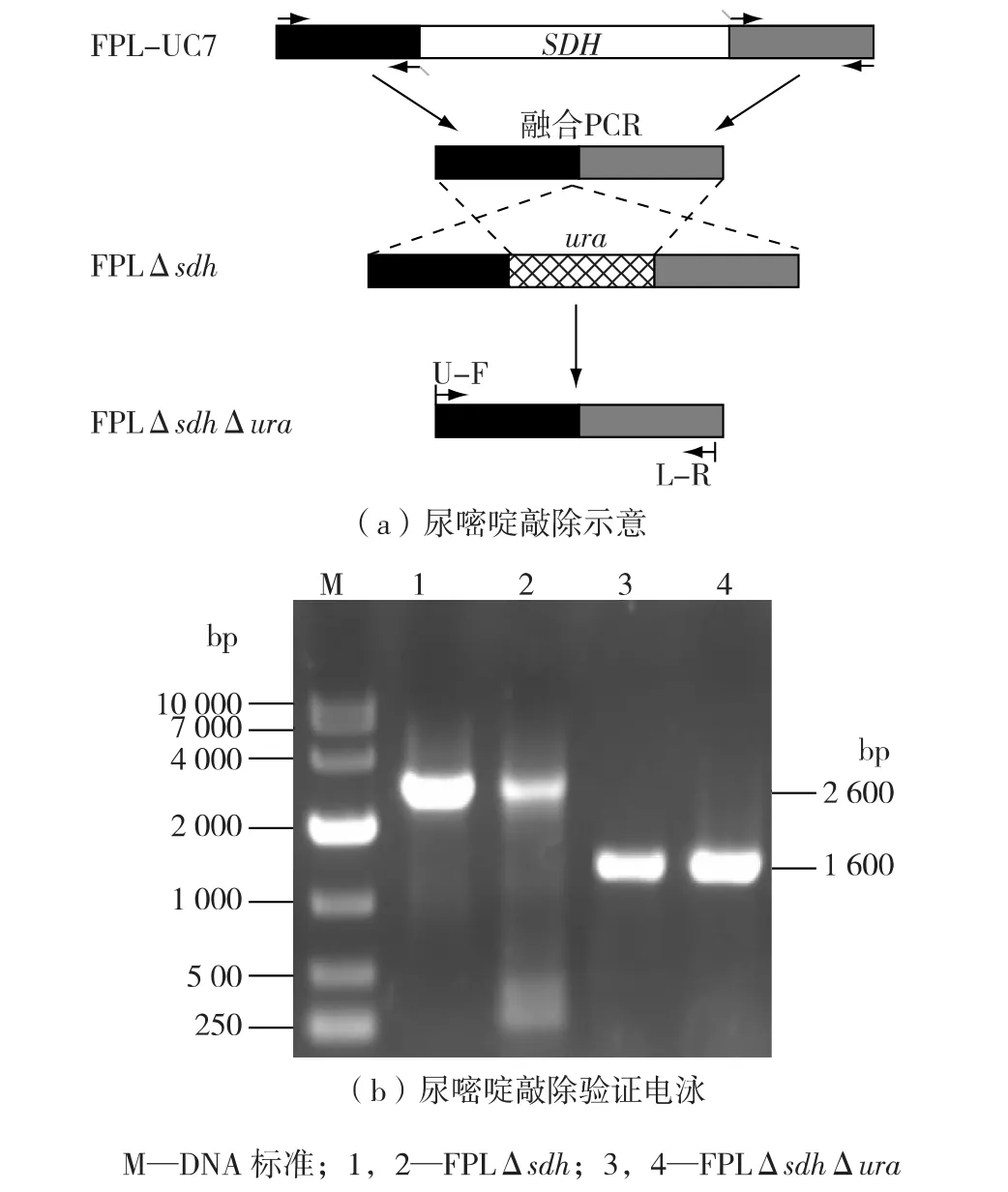
图4 尿嘧啶敲除示意和敲除验证电泳Fig.4 Disruption of ura gene with homologous recombination and PCR verification
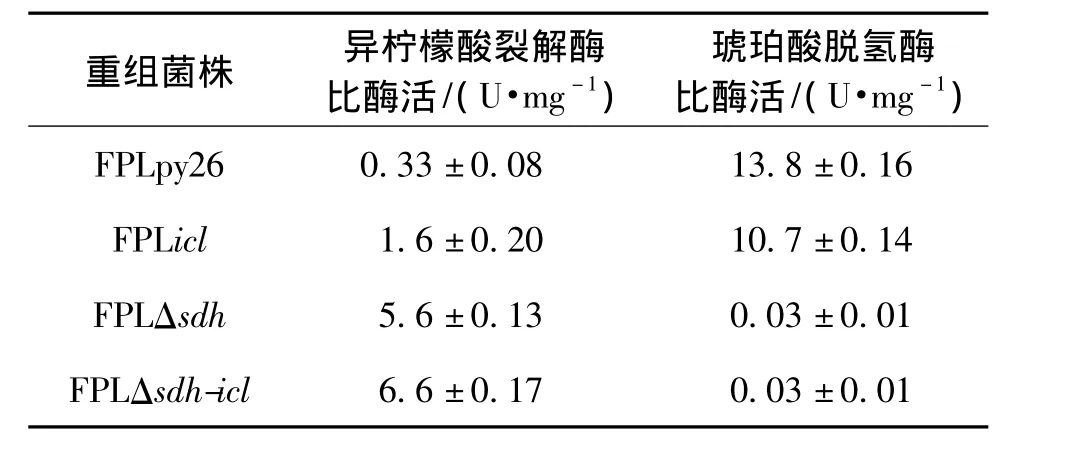
表2 重组菌株和出发菌株中关键酶活性的测定Table 2 Specific activities of key enzymes in P.stipitis strains
2.4 重组菌株发酵生产琥珀酸
将重组菌株 FPLicl、FPLΔsdh、FPLΔsdh-icl和出发菌株FPLpy26于5 L发酵罐中进行培养,进行3次重复实验,其生产琥珀酸的发酵过程曲线如图5所示,相关发酵参数列于表3。由表3可知:①与出发菌株相比,菌株FPLicl耗糖速率和菌株生长没有发生变化,而发酵时琥珀酸质量浓度为0.30 g/L,提高了6倍。副产物乙酸质量浓度则从5.30 g/L(对照菌株)降为2.50 g/L,降低了53%。其原因在于icl1基因的过量表达,导致乙醛酸代谢流量增大,丙酮酸代谢支路流量减少;②菌株FPLΔsdh细胞生长并不受sdh1基因删除所影响,但与出发菌株相比,琥珀酸质量浓度达到1.2 g/L,糖耗速率降低26%,而副产物乙酸质量浓度则增加了105%(10.9 g/L)。其原因在于sdh1基因缺失后TCA循环受阻,菌株糖代谢能力降低,与此同时,异柠檬酸裂解酶比酶活增大(表2),导致乙醛酸循环代谢流量增大,乙酸合成途径也成为重要的产能途径[17];③在缺失sdh1基因的同时过量表达icl1基因使琥珀酸产量达到1.6 g/L,比菌株 FPLicl、FPLΔsdh分别提高了433%和25%;而副产物乙酸则比菌株FPLΔsdh减少30%。胞内乙醛酸流量继续增大,而乙酸代谢途径流量相对减少,从而使琥珀酸产量进一步提高,乙酸浓度降低。

图5 对照菌和重组菌发酵结果比较Fig.5 Comparison of total xylose decrease,biomass,production of succinic acid and acetate between contract strain and recombinant strains
3 结论
借助树干毕赤酵母全基因组代谢网络模型iTL885对琥珀酸最佳合成途径进行设计,发现过量表达icl1基因和敲除sdh1基因能有效地提高琥珀酸的产量。在此基础上,借助代谢工程策略,构建了重组菌株FPLicl(过量表达icl1基因)、FPLΔsdh(敲除sdh1基因)、FPLΔsdh-icl(过量表达 icl1基因和敲除sdh1基因)。重组菌株异柠檬酸裂解酶和琥珀酸脱氢酶比酶活分别为1.6、5.6、6.6 U/mg和10.7、0.3、0.3 U/mg,而琥珀酸产量为0.30、1.20 和1.60 g/L,实现了琥珀酸的初步积累。目前国内外还没有代谢改造树干毕赤酵母生产琥珀酸的相关报道,本文中笔者结合GSMM对树干毕赤酵母进行代谢改造,为以木糖为底物发酵生产琥珀酸奠定了坚实基础。
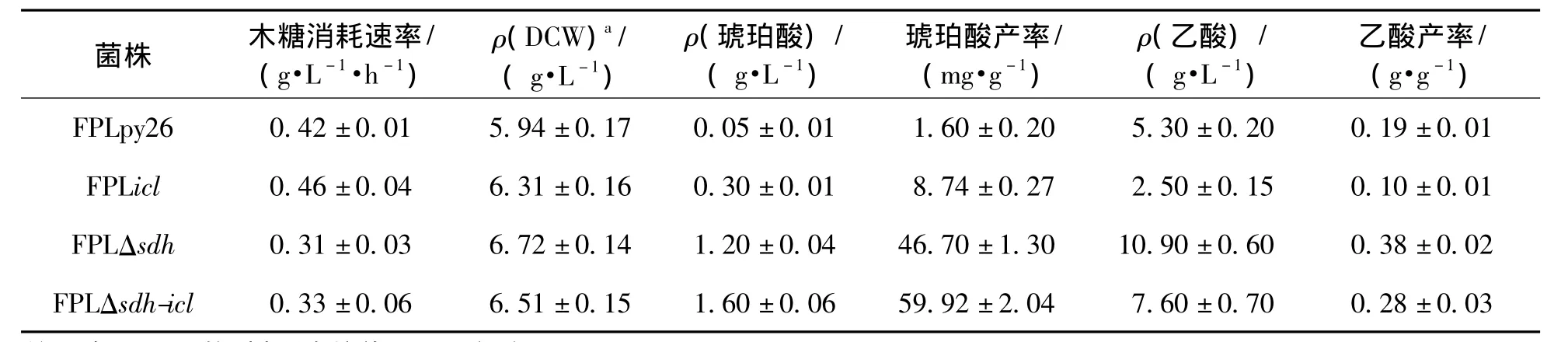
表3 重组菌株和对照菌株发酵参数的比较Table 3 Parameters of P.stipitis shake flask cultures with different strains
[1] Cheng K K,Zhao X B,Zeng J,et al.Biotechnological production of succinic acid:current state and perspectives[J].Biofuel Bioprod Bioresour,2012,6(3):302-318.
[2] Guettler M V,Rumler D,Jain M K.Actinobacillus succinogenes sp.nov.,a novel succinic-acid-producing strain from the bovine rumen[J].Int J Syst Bacteriol,1999,49(1):207-216.
[3] Lee P C,Lee S Y,Hong S H,et al.Isolation and characterization ofa new succinic acid-producing bacterium,Mannheimia succiniciproducens MBEL55E,from bovine rumen[J].Appl Microbiol Biotechnol,2002,58(5):663-668.
[4] Oh I J,Lee H W,Park C H,et al.Succinic acid production from continuous fermentation process using Mannheimia succiniciproducens LPK7[J].J Microbiol Biotechnol,2008,18(5):908-912.
[5] Lee P C,Lee S Y,Chang H N.Succinic acid production by Anaerobiospirillum succiniciproducens ATCC 29305 growing on galactose,galactose/glucose,and galactose/lactose [J].J Microbiol Biotechnol,2008,18(11):1792-1796.
[6] Vemuri G N,Eiteman M A,Altman E.Succinate production in dual-phase Escherichia coli fermentations depends on the time of transition from aerobic to anaerobic conditions[J].J Ind Microbiol Biotechnol,2002,28(6):325-332.
[7] Vemuri G N,Eiteman M A,Altman E.Effects of growth mode and pyruvate carboxylase on succinic acid production by metabolically engineered strainsofEscherichia coli[J].ApplEnviron Microbiol,2002,68(4):1715-1727.
[8] Cheng H Y,Yu R C,Chou C C.Increased acid tolerance of Escherichia coli O157:H7 as affected by acid adaptation time and conditions of acid challenge[J].Food Res Int,2003,36(1):49-56.
[9] Hippe H,Hagelstein A,Kramer I,et al.Phylogenetic analysis of Formivibrio citricus, Propionivibrio dicarboxylicus,Anaerobiospirillum thomasii,Succinimonas amylolytica and Succinivibrio dextrinosolvens and proposal of Succinivibrio naceae fam.nov[J].Int J Syst Bacteriol,1999,49(2):779-782.
[10] Tee W,Korman T M,Waters M J,et al.Three cases of Anaerobiospirillum succiniciproducens bacteremia confirmed by 16S rRNA gene sequencing[J].J Clin Microbiol,1998,36(5):1209-1213.
[11] Slininger P J,Bothast R J,Ladisch M R,et al.Optimum pH and temperature conditions for xylose fermentation by Pichia stipitis[J].Biotechnol Bioeng,1990,35(7):727-731.
[12] Liu T,Zou W,Liu L,et al.A constraint-based model of Scheffersomyces stipitisforimproved ethanolproduction[J].Biotechnol Biofuels,2012,5(1):72.
[13] Lu P,Davis B P,Hendrick J,et al.Cloning and disruption of the beta-isopropylmalate dehydrogenase gene(LEU2)of Pichia stipitis with URA3 and recovery of the double auxotroph[J].Appl Microbiol Biotechnol,1998,49(2):141-146.
[14] Li A,Liu Z,Li Q,et al.Construction and characterization of bidirectional expression vectors in Saccharomyces cerevisiae[J].Fems Yeast Res,2008,8(1):6-9.
[15] Orth J D,Thiele I,Palsson B O.What is flux balance analysis?[J].Nature Biotechnol,2010,28(3):245-248.
[16] Wu H,Li Z M,Zhou L,et al.Improved succinic acid production in the anaerobic culture of an Escherichia coli pflB ldhA double mutant as a result of enhanced anaplerotic activities in the preceding aerobic culture[J].Appl Environ Microbiol,2007,73(24):7837-7843.
[17] Raab A M,Gebhardt G,Bolotina N,et al.Metabolic engineering of Saccharomyces cerevisiae for the biotechnological production of succinic acid[J].Metab Eng,2010,12(6):518-525.
猜你喜欢
中国民间疗法(2021年18期)2021-11-02 08:20:34
核农学报(2020年2期)2020-03-11 08:33:16
食品与发酵工业(2018年3期)2018-04-12 09:35:51
浙江工业大学学报(2017年5期)2018-01-22 02:03:43
菏泽学院学报(2017年2期)2017-05-16 08:59:11
广东饲料(2016年1期)2016-12-01 03:43:01
工业微生物(2016年5期)2016-11-11 06:58:44
化学与生物工程(2016年10期)2016-11-10 06:01:35
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:18
陕西师范大学学报(自然科学版)(2015年1期)2016-01-16 03:23:30
