
再谈催化剂改变化学平衡
2014-04-27 10:43贺传正
沈阳大学学报(自然科学版) 2014年2期
贺传正
(沈阳大学,辽宁 沈阳 110044)
十几年前,作者提出了催化剂改变化学平衡的观点[1].本文从实验、体系的相关计算和理论分析出发,阐述了催化剂对化学平衡状态的影响.
1 实 验
本文的实验为:在0.5mol·L-1的 H2SO4介质 中,Ce(SO4)2滴 定 H3AsO3(E+=1.44V,EAsO4/H3AsO3=0.58V)、Ce(SO4)2滴定 KI(E=0.63V)和I2滴定 H3AsO3的实验.式中,符号○-代表标准状态,即温度为0℃,压力为1大气压(101.325kPa).在化学计量点时,Ce,As,I的浓度为cCe=1.00×10-3mol·L-1,cAs=5.00×10-4mol·L-1,cI=1.00×10-3mol·L-1.实验以饱和甘汞电极作参比电极,以铂作指示电极,在电磁机的搅拌下,每次滴加0.100 mL滴定剂,记录PXS-20型离子活度计(上海第二分析仪器厂生产)显示的稳定毫伏数,把测得的电位值换算成相对于氢标准电极的电位值,绘制出如文献[2]中所示的Ce(SO4)2滴定 H3AsO3+KI体系的滴定曲线.
2 体系的相关计算
由标准状态的条件电极电位E○-,计算不同实验体系化学反应的摩尔自由能变化ΔG○-m、化学计量点的电极电位E和相关组分的平衡浓度.
2.1 Ce(SO4)2 滴定H3AsO3
Ce4+与 H3AsO3的反应速率慢,反应平衡式为
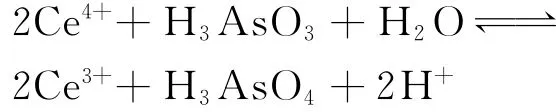
反应的摩尔自由能变化为
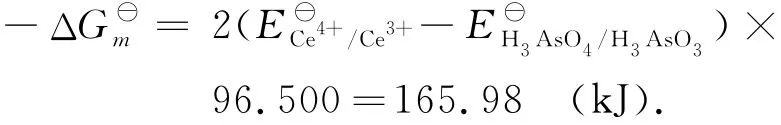
在化学计量点时,[Ce4+]=2[H3AsO3],[Ce3+]=2[H3AsO4],体系的电极电位
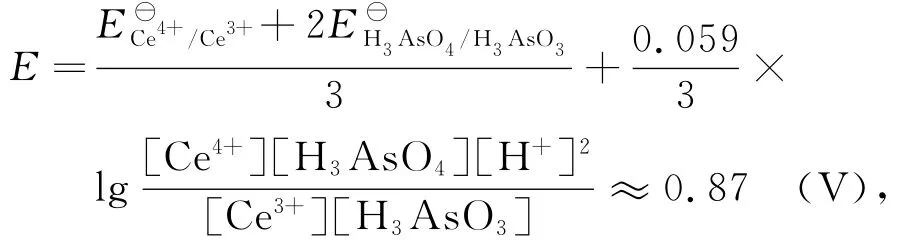
则 [Ce4+]=10-12.72mol·L-1,[H3AsO3]=10-13.02mol·L-1.
2.2 Ce(SO4)2 滴定KI
Ce4+与I-的反应速率快,反应平衡式为

反应的摩尔自由能变化为
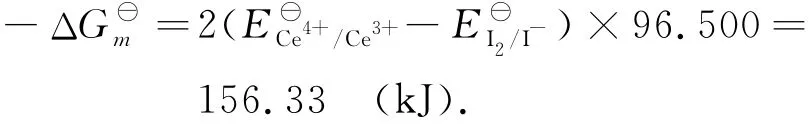
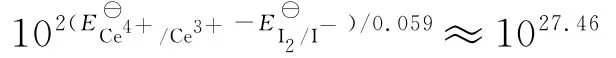
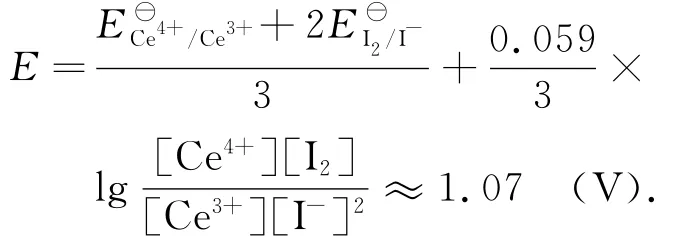
2.3 I2滴定H3AsO3
I2和H3AsO3的反应速率是快的,反应平衡式为

反应的摩尔自由能变化为
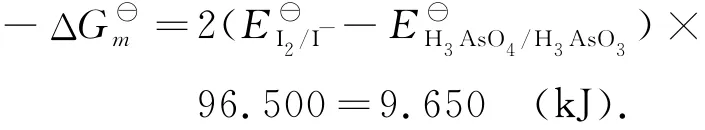
在化学计量点时,[I-]=2[H3AsO4]≈1.00×10-3mol·L-1,[I2]=[H3AsO3],电极电位

此时,[I2]=[H3AsO3]=10-5.50mol·L-1.
2.4 Ce(SO4)2 滴定H3AsO3+KI
I-作催化剂时,体系的反应速率是快的.如果把Ce4+与H3AsO3的反应视为主反应,那么平衡常数
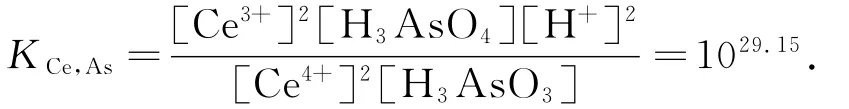
把Ce4+和I-的反应作为副反应,平衡常数
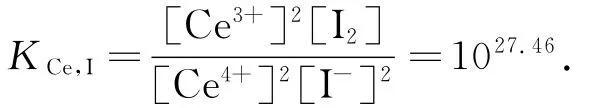
若用[Ce′]表示溶液中游离的Ce4+的浓度以及与I-副反应时消耗的Ce4+的浓度的和,则
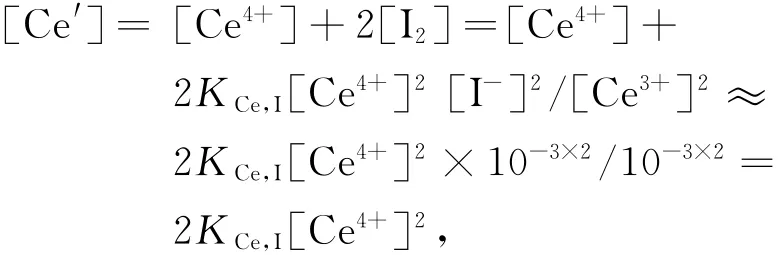
从 而 [Ce4+]2= [Ce′]/2 KCe,I.因 为 [Ce′]=2[H3AsO3],将[Ce4+]2=[H3AsO3]/KCe,I代入KCe,As的式子,则在化学计量点时

电极电位
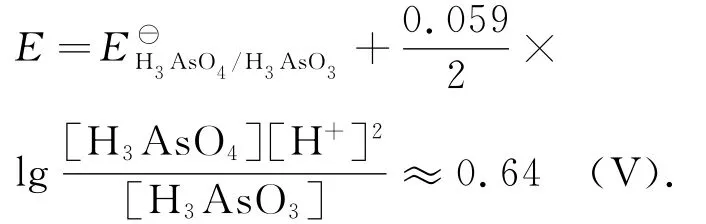
[I2]=[H3AsO3].这些数值与I2滴定 H3AsO3体 系 在 计 量 点 时 的 相 同,但 [Ce4+]为10-16.48mol·L-1.显然,I-和I2都参与到体系的平衡之中.
比较Ce4+滴定H3AsO3的I-催化体系和非催化体系在化学计量点的电极电位、组分的平衡浓度,不难发现二者体系的平衡状态是不同的,即催化剂改变了化学平衡.
3 理论分析
3.1 热化学反应方程式和化学反应平衡式
3.2 盖斯定律的适用条件
盖斯定律认为:“化学反应不管是一步完成还是分几步完成,其反应的焓变或反应的热力学能变相同.”就是说,反应的热力学能变是由反应的始态和终态确定的,与过程经历的途径无关.显然,这里的化学反应是热化学反应,是用热化学反应方程式表示的.盖斯定律的每个分步反应是独自进行的,是变化过程中的一个步骤,而且每步反应的反应物又必须完全转变成反应产物.所以,盖斯定律适用的物质条件是,每步反应的反应物都要完全转变成反应产物.
3.3 催化体系的状态变化不适用盖斯定律
催化反应是连续的可逆反应,每步反应的反应物并不能完全转变成反应产物,所以,不能把每一步反应看作体系变化过程的一个步骤,即不能把每一步反应的平衡式看成一个独立的热化学反应方程式.分步反应平衡式加和得到的反应平衡式也不是总的热化学反应方程式,也就不能用它来表示体系变化的始态和终态,以及相关热力学函数变化值的计算,所以,不能用盖斯定律确定这种体系的状态变化.例如,不能把I-催化Ce4+以及氧化H3AsO3的分步反应平衡式
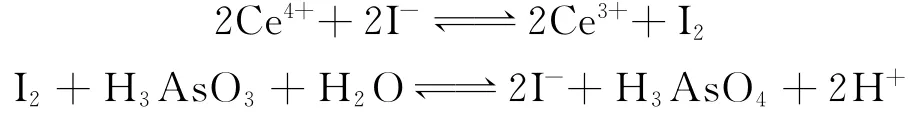
看作体系变化过程的步骤,分步反应平衡式加和得到的反应平衡式
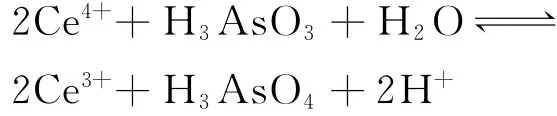
也不能表示体系变化的始态和终态;体系的自由能变化也不等于Ce4+和I-热化学反应的自由能变化(-156.33kJ)与I2和 H3AsO3热化学反应的自由能变化(-9.650kJ)之和(-165.98kJ),尽管这个数值和Ce4+与H3AsO3热化学反应的自由能变化相同.
3.4 催化剂改变化学性质
如果催化剂参与反应,反应后质量和化学性质不改变,体系和环境的状态也没有变化,那么催化剂在反应过程中的状态变化就应该适用盖斯定律.经过一个循环反应过程,自由能变化的封闭积分等于0,那么,催化剂的变化过程是可逆的,实际上,催化剂的变化过程却是自发的,即与热力学第二定律相悖.这正是催化剂在反应过程中不可能完全恢复到原来的化学形态的佐证.相反,如果反应后催化剂完全恢复到原来的化学形态,那么,催化反应就不是可逆的了,那也就无所谓化学平衡了.
3.5 能量最低原理
自发过程的方向是体系由高势能状态向低势能状态变化.催化反应是连续反应,随着反应逐级进行,体系的自由能也逐级下降,当达到末级反应时降到最低,那么,体系的平衡状态就应该由末级的催化反应的自由能的变化来确定.例如,在I-催化Ce4+以及氧化H3AsO3的反应过程中,体系的自由能下降到I2氧化H3AsO3时最低,体系的平衡状态也就由这步反应的自由能的变化确定,即是由Ce4+和H3AsO3反应的自由能变化与Ce4+和I-反应的自由能变化之差确定的,而不是由Ce4+和I-反应的自由能变化与I2和H3AsO3反应的自由能变化之和确定的.那么,I2和 H3AsO3反 应 的 平 衡常数 KI2,As[3]应 表示为
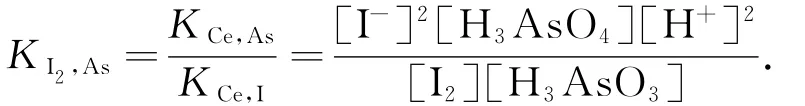
在化学计量点时,[I2]=[H3AsO3].在本文所述的体系中

并且,[I2]=10-5.50mol·L-1,[Ce4+]=10-16.48mol·L-1,E=0.64V.这些数值和用副反应处理体系的平衡关系时相应的计算值相同,但此处是近似认为I-在第一步催化反应中完全被Ce4+氧化成I2,不过误差不会太大.
本文中,Ce4+氧化H3AsO3的I-催化体系和非催化体系在达到计量点时的自由能的变化有差异,可以向后者在计量点时加入KI,伴随着Ce4+和H3AsO4对I-的氧化,体系的电位由0.87V下降到0.64V,计算此过程体系的自由能的变化.
图1中所示的滴定曲线的前半部分是滴入的Ce4+氧化I-为I2,I2被 H3AsO3还原成I-,I-又被Ce4+氧化成I2,直到化学计量点时曲线产生了突跃,由此时消耗的滴定剂Ce4+的量可以计算H3AsO3的含量.这段曲线和用I2滴定H3AsO3的曲线的变化一样.曲线的第二个突跃出现在Ce4+和I-反应的计量点.
滴定曲线显示了I-对Ce4+氧化H3AsO3的拉平效应,使体系的自由能降到最低.

图1 用Ce(SO4)2滴定H3AsO3+KIFig.1 Titrating H3AsO3+KI with Ce(SO4)2
3.6 催化剂不改变非催化反应的活化能
在催化体系中,既有非催化反应,又有连续催化反应,而且,相比之下,非催化反应的速率小得忽略不计.催化剂的存在并没有改变非催化反应的属性,其活化能和速率常数依旧,这可从催化体系和非催化体系的化学计量点及其相应组分的平衡浓度均不相同足以说明,所以,把催化体系反应速率增加的原因归结为降低了非催化反应的活化能是令人费解的.催化体系的反应速率应由催化反应中活化能最高的那步反应确定.
3.7 催化剂对平衡的移动不会太大
作为强度性质,如催化剂的电极电位,它只能处于非催化反应的两个电对的电极电位之间,否则,催化剂在反应中就不能起到承上启下的作用了.所以,催化体系和非催化体系的平衡状态和能量改变的差别不会太大,何况催化剂的加入量往往又很少,平衡状态的改变就更不明显了.
4 结 语
催化剂能改变化学平衡.催化体系的平衡状态由末步的催化反应确定.向处于平衡状态的体系中加入催化剂,平衡能产生移动.催化体系的自由能的变化比非催化体系的低.
[1] 贺传正,阎锋.催化剂对化学平衡的影响[J].辽宁大学学报:自然科学版,1999,26(3):268-270.
(He Chuanzheng,Yan Feng.Influence of Catalyst on Chemical Equilibrium[J].Journal of Liaoning University:Natural Science,1999,26(3):268-270.)
[2] 贺传正,姜汉硕,贾铁军.氧化还原连续滴定的理论研究[J].辽宁大学学报:自然科学版,1984(2):60-69.
(He Chuanzheng,Jiang Hanshuo,Jia Tiejun.A Study of Oxidation Reduction Continuous Titration Theory[J].Journal of Liaoning University:Natural Science,1984(2):60-69.)
[3] 贺传正.共存组分参与的反应的平衡处理[J].沈阳大学学报,2000,12(4):28-31.
(He Chuanzheng.Balance Treatment of the Reaction with Coexisting Component [J]. Journal of Shenyang University,2000,12(4):28-31.)
猜你喜欢
食品安全导刊(2021年20期)2021-11-28
小星星·阅读100分(高年级)(2020年8期)2020-10-20
民间故事选刊·上(2020年6期)2020-06-29
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
小小说月刊(2019年1期)2019-01-24
中学化学(2017年6期)2017-10-16
电镀与环保(2016年2期)2017-01-20
中学化学(2016年10期)2017-01-07
