
口炎清胶囊的质量控制和稳定性试验
2014-04-27 08:45:15杨秋凤
中国药业 2014年11期
江 涛,雷 健,邢 茂,杨秋凤,葛 勤
(中国人民解放军第三军医大学新桥医院药剂科,重庆 400037)
扁平苔藓是发生于口腔黏膜的一种非感染慢性炎症,病损难愈,给患者造成严重的心理压力[1-2]。我院研制的由赤芍、黄连、麦冬等药材制成的中药汤剂,具有解毒消肿、滋阴清热等作用,临床用于扁平苔藓等口腔炎症,疗效较好;但汤剂口感不好,患者服药依从性差,且水溶液久贮易酸败,不易携带,应用受到一定限制。为解决上述问题,笔者将中药汤剂制成胶囊。胶囊剂为固体制剂,有效期相对较长,易携带,患者依从性较好。为保证制剂质量,本试验中对口炎清胶囊的制备工艺、质量控制和稳定性进行了研究。
1 仪器与试药
1200型高效液相色谱仪仪系统(美国Agilent公司);HWS24型电热恒温水浴锅(上海科技一恒有限公司);KQ5200型超声波清洗器(昆山市超声仪器有限公司);电热鼓风干燥箱(重庆市永生实验仪器厂);电子天平(赛多利斯科学仪器北京有限公司);SHH-SDT型综合药品稳定性试验箱(重庆市永生实验仪器厂)。盐酸小檗碱(批号为110713-200911)、黄芩苷(批号为110715-201117)、芍药苷(批号为 110736-201136)对照品,均购自中国食品药品检定研究院;黄连、黄芩、麦冬、赤芍等中药材均购自重庆桐君阁中药批发有限公司,经重庆医科大学药学院刘新教授鉴定为真品;淀粉(曲阜市药用辅料有限公司,批号为20100201);硬脂酸镁(安徽山河药用辅料有限公司,批号为110916);口炎清胶囊(自制,批号为 20120109,20120116,20120120);乙腈为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 制备
取处方中全部中药材,80%乙醇提取3次,每次2 h,合并提取液,回收乙醇,浓缩,70℃烤成干浸膏。干浸膏粉碎,过80目筛,细粉加入20%淀粉,混匀,90%乙醇制成软材,制粒,湿颗粒于60℃干燥3 h,20目筛整粒,得干颗粒。干颗粒加1%硬脂酸镁,混合,0号胶囊填充,即得口炎清胶囊。
2.2 一般质量控制项目
性状:本品为胶囊剂,内容物为棕红色至棕色颗粒,气香,味微苦。
水分测定:照 2010 年版《中国药典(一部)》附录ⅨH[3]中第一法(烘干法)测定。结果3批口炎清胶囊(批号分别为20120109,20120116,20120120)的含量水分别为 4.7% ,4.2% ,5.1% ,均小于9.0%,符合药典规定。
装量差异差异测定:照 2010 年版《中国药典(一部)》附录ⅠL[3]10中胶囊剂装量差异项下方法测定。结果3批口炎清胶囊(批号分别为20120109,20120116,20120120)的装量差异分别为±6.3%,±5.5%,±5.3%,均小于10%,符合药典规定。
崩解时限测定:照 2010 年版《中国药典(一部)》附录Ⅻ A[3]71中崩解时限检查法胶囊剂项下方法测定。结果3批口炎清胶囊(批号分别为 20120109,20120116,20120120)的崩解时限分别为 20,23,21 min,均在 30 min内,符合药典规定。
2.3 薄层色谱法定性鉴别
黄连:取口炎清胶囊内容物2g,加甲醇25mL,加热回流15min,滤过,滤液补加甲醇至25mL,作为供试品溶液。另取黄连对照药材0.25 g,同法制成对照药材溶液。再取盐酸小檗碱对照品,加甲醇制成每1mL含0.5 mg的溶液,作为对照品溶液。再按处方比例,取缺黄连的其他各味药材,按制备工艺加工制成阴性样品,按供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法[3]34试验,吸取上述4种溶液各1μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯-异丙醇-甲醇-水-三乙胺(3∶3.5∶1∶1.5∶0.5∶1)为展开剂,置氨蒸气饱和的展开缸内,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液、对照品溶液色谱相应位置上,显相同颜色的荧光斑点,阴性无干扰,见图1 A。
麦冬:取口炎清内容物1 g,加盐酸0.5mL、水10mL,加热煮沸5min,放冷,三氯甲烷提取3次,每次7mL,分取三氯甲烷液,浓缩至1mL,作为供试品溶液。另取麦冬对照药材1 g,同法制成对照药材溶液。取缺麦冬的其他各味药材,按制备工艺加工制成阴性样品,按供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法试验,吸取上述3种溶液各5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-丙酮(4∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至墨绿色斑点显色清晰。供试品溶液色谱中,在与对照药材溶液色谱相应位置显相同颜色的斑点,阴性对照品溶液无干扰,见图1 B。
黄芩:取本品3 g,加水40mL,超声30min,加乙醚振摇提取2次,每次50mL,弃乙醚液,剩余溶液用乙酸乙酯振摇提取3次,每次40mL,合并乙酸乙酯液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。另取黄芩对照药材0.5 g,同法制成对照药材溶液。再取黄芩苷对照品,加甲醇制成每1mL含1mg的溶液,作为对照品溶液。取缺黄芩的其他各味药材,按制备工艺加工制成阴性样品溶液,按供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法试验,吸取上述3种溶液各5μL,分别点于同一硅胶G薄层板上,以乙酸乙酯 -丙酮 -醋酸 -水(10∶4∶5∶3)为展开剂,预平衡30min,展开,取出,晾干,喷以5%三氯化铁乙醇溶液。供试品色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液无干扰,见图1 C。

图1 薄层色谱图
2.4 芍药苷含量测定[4-6]
2.4.1 色谱条件与系统适用性试验
色谱柱:HypersilODS柱(150mm×4.6mm,10μm);流动相:乙腈 -0.1%磷酸溶液(13∶87);检测波长:229 nm;流速:1mL /min;柱温:室温;进样量:20μL。理论板数按芍药苷峰计算应大于3000。
2.4.2 溶液制备
精密称取芍药苷对照品12.5mg,置25mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得对照品溶液。取口炎清浸膏粉2 g,精密称定,置50mL容量瓶中,加40mL甲醇超声处理30min,冷却至室温,加甲醇至刻度,振摇,过滤,即得供试品溶液。取缺赤芍的其他各味药材,80%乙醇提取3次,每次2 h,合并提取液,浓缩,70℃烤成干浸膏,粉碎,按供试品溶液制备方法制备缺赤芍的阴性对照品溶液。
2.4.3 方法学考察
专属性试验:取2.4.2项下3种溶液,分别进样20μL,按拟订色谱条件依法进样测定,记录色谱图。结果阴性对照品溶液对芍药苷的测定无干扰,见图2。
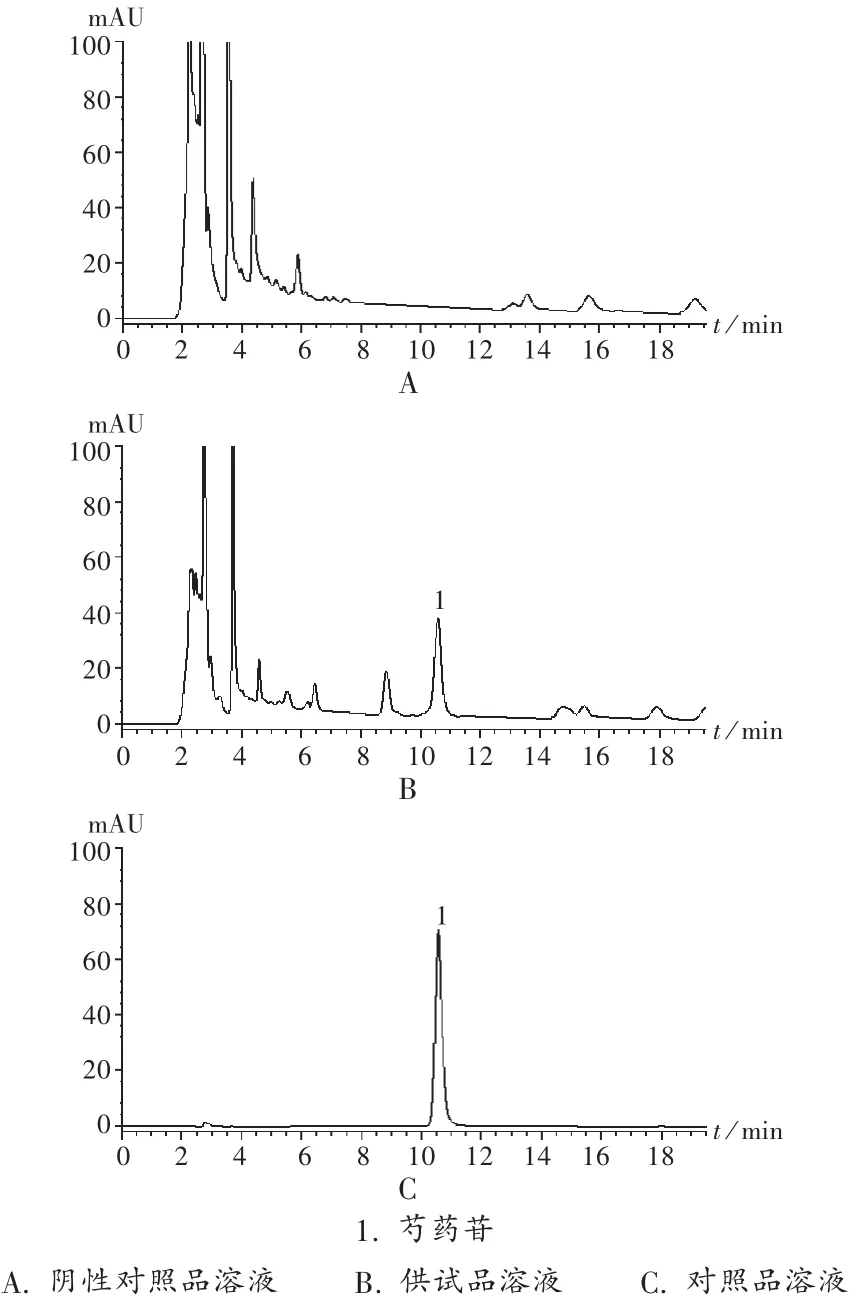
图2 高效液相色谱图
线性关系考察:精密吸取0.5 g/L芍药苷对照品溶液0.1,0.5,1.5,2.5,4.5mL,置 5mL 容量瓶中,甲醇稀释至刻度,摇匀,制成质量浓度分别为 0.05,0.15,0.25,0.35,0.45 g/L 的溶液。分别吸取20μL,按拟订色谱条件进样测定,记录峰面积。以质量浓度为横坐标(X)、峰面积(Y)为纵坐标进行线性回归,得回归方程Y=13997X-52.45(r=0.9999)。结果表明,芍药苷质量浓度在0.05~0.45 g/L范围内与峰面积呈良好线性关系。
精密度试验:精密吸取芍药苷对照品溶液20μL,按拟订色谱条件重复进样6次,测定,结果芍药苷峰面积的RSD为0.75%(n=6);连续测定4 d,结果芍药苷峰面积的RSD为0.68%(n=6),表明仪器精密度良好。
重复性试验:取口炎清浸膏粉5份,依法制备供试品溶液,精密吸取20μL,按拟订色谱条件测定,记录峰面积。结果芍药苷峰面积的RSD为1.15%(n=5),表明方法重复性良好。
稳定性试验:精密吸取同一供试品溶液20μL,按拟订色谱条件,于 0,2,4,8,24 h 时进样,依法测定并记录峰面积。结果芍药苷峰面积的RSD为0.88%(n=5),表明供试品溶液在24 h内稳定。
加样回收试验:取已知芍药苷含量的口炎清浸膏粉0.1 g共9份,分别置10mL容量瓶中,分别加入0.5 g/L的对照品溶液1.0,1.2,1.5mL,制成低、中、高 3 种质量浓度,分别精密吸取 20μL,按拟订色谱条件进样测定,计算回收率。结果见表1。2.4.4 含量测定
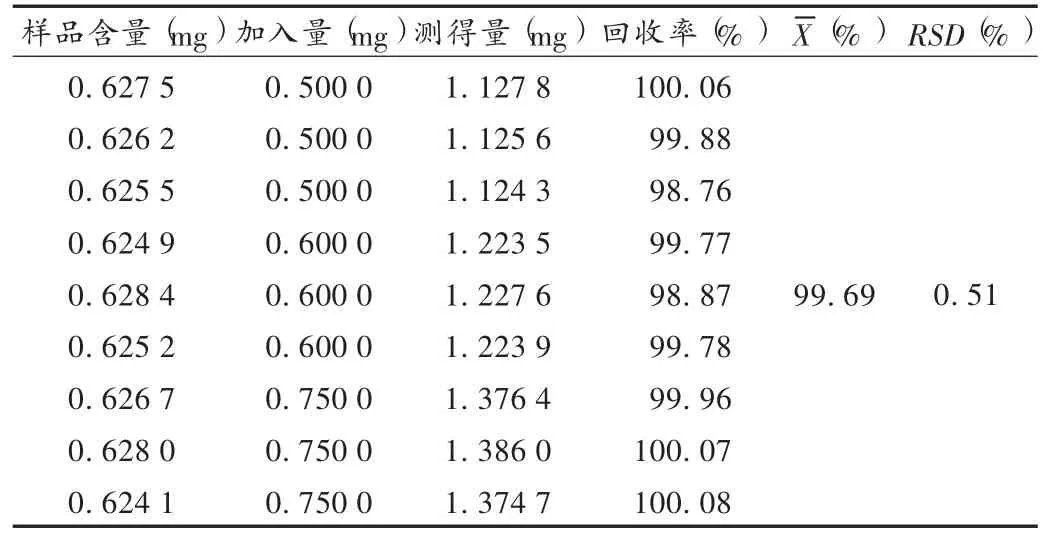
表1 芍药苷加样回收试验结果(n=9)
分别取3批口炎清胶囊,依法制备样品溶液,按拟订色谱条件,精密吸取样品溶液20μL,进样测定芍药苷含量。结果见表2。
2.5 稳定性考察[7]
2.5.1 影响因素试验
高温试验:取口炎清胶囊(批号为20120109),置平皿中,60℃恒温箱中放置10 d时,于第5,10天时取样,测定水分、崩解时限、芍药苷含量。高湿度试验:取口炎清胶囊(批号为20120109),置平皿中,在25℃ 、相对湿度(75±5)%的恒湿密闭容器中放置10 d,于第5,10天时取样,测定水分、崩解时限、芍药苷含量。强光照射试验:取口炎清胶囊(批号为20120109),置平皿中,放在装有日光灯的光照箱内,于照度(4500±500)lx条件下放置10 d,于第5,10天时取样,观察性状,测定水分、崩解时限、芍药苷含量。结果在上述试验条件下口炎清胶囊内容物为棕红色至棕色颗粒,气香,味微苦,其他检查项目均符合要求,见表3。2.5.2 加速试验
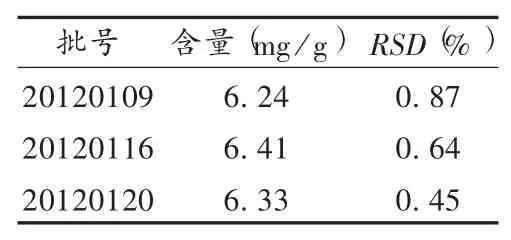
表2 口炎清胶囊中芍药苷含量测定结果(n=3)
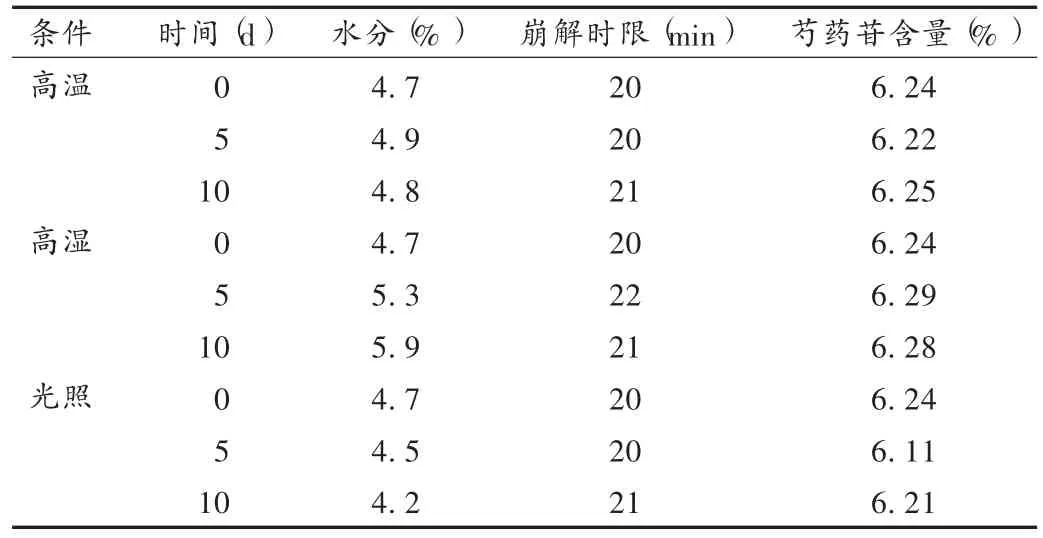
表3 口炎清胶囊的影响因素试验结果
取 3批口炎清胶囊(批号分别为 20120109,20120116,20120120),模拟临床用药包装(口服药用高密度聚乙烯瓶,铝箔封口,瓶内放干燥剂),在温度(40±2)℃ 和相对湿度(75±5)%条件下放置6个月,在试验第0,1,2,3,6个月末分别取样,测定水分、崩解时限、芍药苷含量。结果口炎清胶囊内容物为棕红色至棕色颗粒,气香,味微苦,其他检查项目均符合要求,见表4。
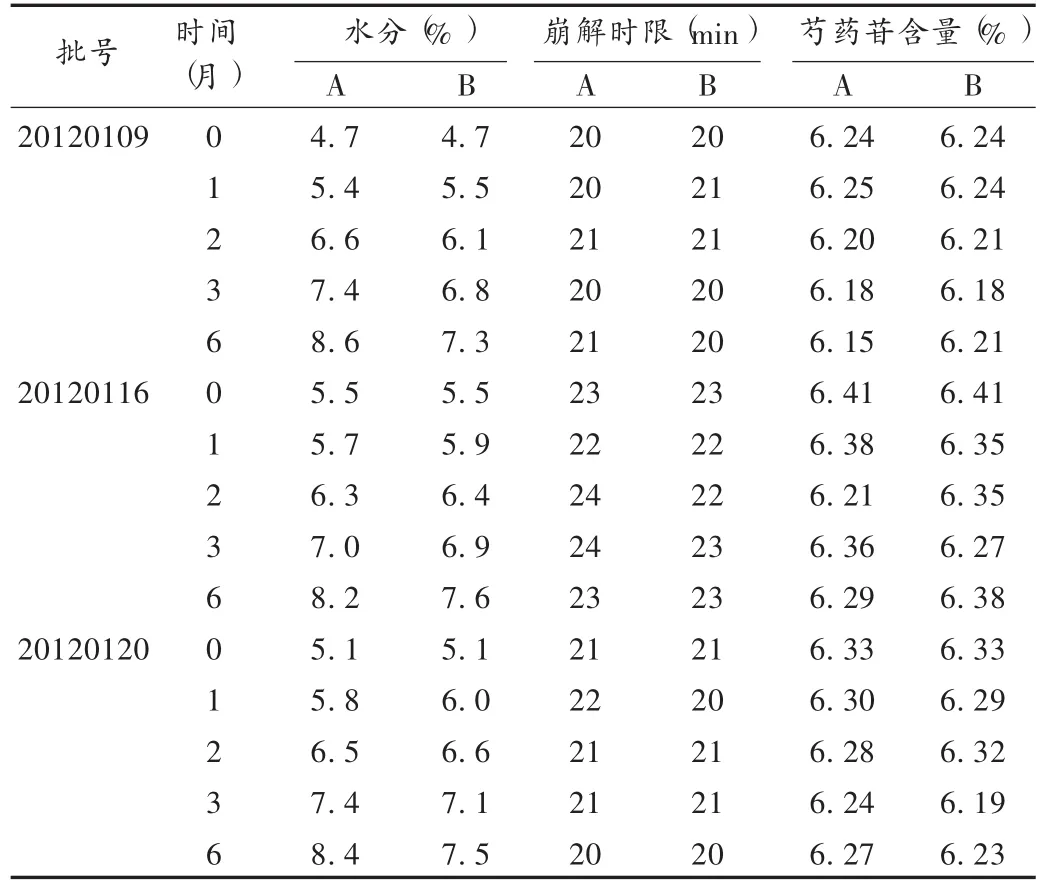
表4 口炎清胶囊的加速试验和长期试验结果
2.5.3 长期试验
取 3批口炎清胶囊(批号分别为 20120109,20120116,20120120),模拟临床用药包装(口服药用高密度聚乙烯瓶,铝箔封口,瓶内放干燥剂),在温度(25 ±2)℃,相对湿度(60 ±10)%条件下放置 12 个月,分别于 0,3,6,9,12 个月时取样,测定水分、崩解时限、芍药苷含量。结果口炎清胶囊内容物为棕红色至棕色颗粒,气香,味微苦,其他检查项目均符合要求,见表4。
3 讨论
药材产地影响药材的品质,药材品质直接影响制剂的质量,产地不同的药材有效成分含量均有差异[8-9]。同一产地的药材,也会因采收时间、贮藏、运输等不同,质量差异很大[9-10]。因此,在制订制剂质量标准时,首先需对药材质量进行严格控制,保证药材符合2010年版《中国药典》的有关规定,这是保证制剂质量的先决条件。
黄芩的鉴别首先参考2010年版《中国药典(一部)》黄芩项下的薄层色谱鉴别方法,但由于本方为复方制剂,成分复杂,样品点样困难,色谱斑点分离不好,背景干扰较大。因此,通过多次试验,不断摸索提取方法,调节展开条件及显色方法,结果所用方法分离良好,斑点清晰,简便易行,重复性好,可作为该制剂的定性质量控制方法。
本试验比较了多种流动相,如乙腈-0.02moL/L磷酸二氢钠、乙腈-0.1%磷酸、乙腈-水、甲醇-水、乙腈-0.5%三乙胺水、甲醇-0.4%磷酸、甲醇-0.05moL/L磷酸二氢钾的分离效果。结果,乙腈-0.1%磷酸的分离效果最为理想,芍药苷色谱峰的保留时间适当,分离度高,重现性好。
参考文献:
[1]张延琳,丁 蕾,逄爱惠,等.刺五加黄芪片治疗口腔扁平苔藓患者的临床观察[J]. 中国医院药学杂志,2008,28(13):1091 -1093.
[2]常 珍,李容林,李春阳.细胞自噬在口腔扁平苔藓恶变中作用的研究进展[J]. 国际口腔医学杂志,2012,39(3):416 -420.
[3]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:附录52.
[4]李中娥.HPLC法同时测定妇科分清丸中阿魏酸和芍药苷[J].中成药,2012,34(3):487 -489.
[5]高 辉,马小军,林永强,等.健脑补肾丸定性定量方法的研究[J].时珍国医国药,2012,23(1):176 -178.
[6]王 芳,胡雪莲,陈 琳.高效液相色谱法测定胃灵胶囊中芍药苷含量[J]. 中国药业,2011,20(1):17 -18.
[7]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:附录199-201.
[8]何恩慧,夏 洋.浅述中药材对临床疗效与质量的关系[J].中国现代药物应用,2011,5(2):233 -234.
[9]李卫平,邹 龙.我国中药材资源保护现状及发展对策[J].中国药业,2011,20(20):5 - 6.
[10]吴志利,李 璨,游 娟.发展我国中药材物流的建议[J].中国药房,2011,22(47):4417 - 4419.
猜你喜欢
今日农业(2022年13期)2022-09-15 01:18:00
上海文化(文化研究)(2021年6期)2022-01-07 10:30:46
今日农业(2021年21期)2021-11-26 05:07:00
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
中成药(2019年12期)2020-01-04 02:02:44
北方牧业(2016年1期)2016-12-17 19:08:50
火花(2015年3期)2015-02-27 07:40:49
中国卫生标准管理(2015年8期)2015-01-26 18:08:35
中国中医药现代远程教育(2014年20期)2014-03-01 04:31:29
当代畜禽养殖业(2014年7期)2014-02-27 07:59:18
