
XAFS法研究煤灰熔融过程中Fe元素形态的变化
2014-04-14 02:24杨俊波范浩杰刘俊杰杨润东张忠孝
动力工程学报 2014年6期
杨俊波, 范浩杰, 刘俊杰, 杨润东, 张忠孝
(上海交通大学 机械与动力工程学院,上海 200240)
煤灰是一种极为复杂的无机混合物,煤灰熔融特性是动力用煤和气化用煤的重要质量指标.由于煤灰熔融机理极其复杂,目前研究手段主要有热重-差热分析仪(TG-DSC)、扫描电镜(SEM),X射线衍射仪(XRD)和傅里叶红外光谱(FT-IR)等[1-2],但这些方法大多停留在宏观物质及矿物质组成层面,难以从元素及原子构成角度观察熔融过程.煤灰在高温熔融过程中,Fe元素的变化对煤灰整体的熔融情况起着至关重要的作用.张德祥等[3]的研究表明,Fe2O3的含量增加大大降低了神府煤灰的熔融温度.Zhang等[4]研究了燃烧条件下铁系矿物质对煤灰熔融特性的影响,结果表明,不同价态的Fe是煤灰在不同条件下呈现不同熔融特性的主要原因,煤灰中的矿物质(如方铁矿、贵榴石和铁橄榄石等)在1 273~1 373 K的温度范围内影响着煤灰在还原性条件下的熔融特性.但是对于熔融过程中Fe的价态及形态是如何变化的,限于实验手段很少有人深入研究.
X射线吸收精细结构 (XAFS,x-ray absorption fine structure)技术利用X射线检测中心原子的K边吸收系数精细结构变化,可以提供配位距离、配位数和近邻配位原子种类等吸收原子的近邻几何结构信息、吸收原子的氧化态和配位化学(如四面体、八面体的配位)等信息,从而得到了其他方法难以得到的结构信息.自从同步辐射光源出现以来,XAFS技术得到了长足的发展,且XAFS法可以研究固态、液态和气体等材料以及几乎所有凝聚态物质的局域结构.因此,XAFS法已被认为是一种强有力的结构探测手段[5-7].
笔者利用高强度的上海同步辐射光源,采用XAFS法对高灰熔点和低灰熔点煤灰样在还原性气氛下熔融过程中Fe的价态变化及变化机制进行了研究,并分析了Fe周围的配位环境.
1 实验部分
1.1 样品的制备
限于实验条件,采用GB/T 212—2008《煤的工业分析方法》中快速制灰法制灰.虽然该方法与实际炉内条件有一些差别,会导致灰成分的一些变化,但目前仍是一种被广泛使用的制灰方法.表1给出了2种实验煤样的灰成分分析.
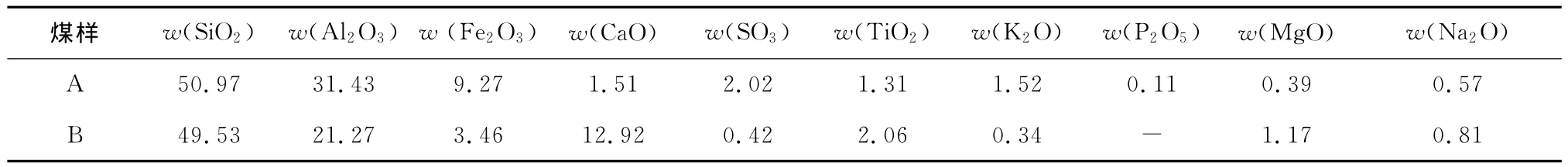
表1 灰成分分析Tab.1 Analysis on ash composition %
在弱还原性气氛(体积流量比qV(CO)∶qV(CO2)=2∶1)下,采用高温热显微镜观察煤灰样的宏观熔融特性,几个熔融特征温度见图1,具体如下:A煤灰样变形温度TDT为1 386℃,软化温度TST为1 424℃,流动温度TFT为1 487℃,为标准的高灰熔点煤灰样;B煤灰样变形温度为1 116℃,软化温度为1 125℃,流动温度为1 213℃,为低灰熔点煤灰样.选用熔点差异较大的这2个煤灰样作为XAFS分析样品,目的是比较不同熔融过程中(如在形成低温共融物和高温共融物过程中)Fe元素形态变化的差异.
将制得的A煤灰样在弱还原性气氛下加热,得到加热到900℃、1 100℃、1 300℃和熔融态的样品,分别记为 A900、A1100、A1300和 A molten state.采用同样的方法处理B煤灰样,得到加热到900℃、1 100℃和熔融态的样品,分别记为B900、B1100和B molten state.

图1 高温热显微镜下煤灰样的熔融过程Fig.1 Ash fusion process under high-temperature microscope
1.2 实验原理及方法
X射线穿透物质后,由于光电效应被物质吸收,其强度减弱.研究发现,物质的X射线吸收系数μ在某些位置出现吸收突越,该位置称之为吸收边(见图2(a)).进一步发现在吸收边附近及高能吸收延伸段存在分离的峰和波状起伏,称之为XAFS.
图2(b)给出了XAFS产生的图像化的解释.其中左半部分横轴表示吸收原子及一个近邻配位原子的一维空间分布,纵轴表示能量,原子势阱处处于核能级能量.处于连续能级的光电子以波的形式向外传播,由于近邻配位原子的存在而产生散射,背散射波与出射波在吸收原子处发生干涉,从而调制了吸收原子处光电子波函数的幅度.图2(b)的右半部分为对应的吸收曲线μ(E),纵轴表示能量,与左半部分标度相同,横轴表示吸收系数.图2(b)左、右2部分的对比表明,在不同能量处,背散射波引起的干涉调制了吸收系数μ(E),形成了XAFS.

图2 XAFS物理原理图Fig.2 Physical diagram of XAFS method
XAFS实验在上海同步辐射光源X射线吸收精细结构谱线站BL14W1上进行.光源采用38 pole wiggler,电子能量为3.5 Ge V,磁场强度为1.2 T,光线接收角为1.5 mrad×0.107 mrad(H×V),光斑大小选择非聚焦模式,晶体模式为Si(111).上海同步辐射光源具有高亮度、高强度和高偏振等优点,可以揭示利用其他光源无法得知的科学秘密.
实验方法如下:对已加热至实验温度的样品研磨后,将其均匀平摊至实验胶带上,遵照BL14W1实验步骤依次对实验样品进行XAFS测试.
2 XAFS实验结果与分析
选用Fe2O3和FeCl2分别作为Fe3+和Fe2+标准能谱,并对不同温度下制得的高灰熔点A煤灰样和低灰熔点B煤灰样采用X射线投射法进行XAFS实验.将得到的数据经过Athena软件处理后获得μ-E 曲线.图3给出了标准样Fe3+(Fe2O3)和Fe2+(FeCl2)的 XAFS图谱.
图3(b)中Fe2+的K吸收边XAFS图谱包含近边区(XANES)和扩展边区(EXAFS)2部分.Fe元素K吸收边值Ek为7 112 e V,近边区为7 082~7 262 e V,而扩展边区为7 263~7 912 e V.XANES对吸收原子氧化态和配位化学环境(如四面体配位和八面体配位)非常敏感,而EXAFS一般用来测定吸收原子的近邻配位原子距离、配位数和原子种类.
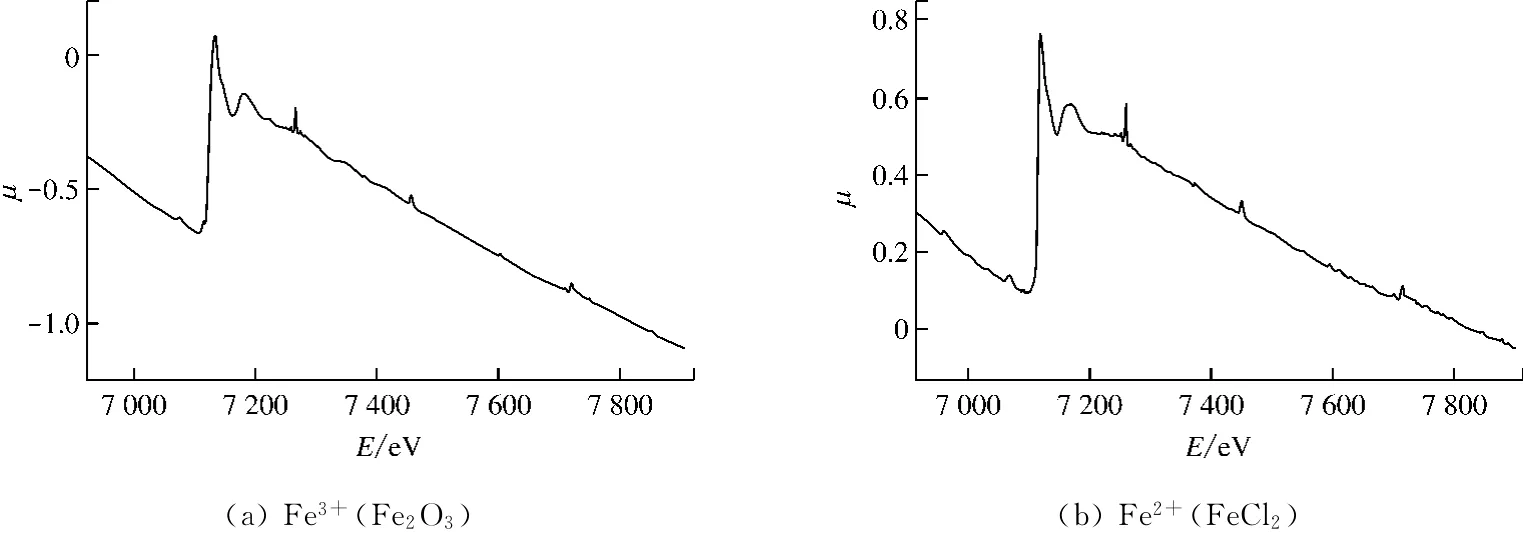
图3 Fe3+(Fe2 O3)和Fe2+(FeCl2)的 XAFS图谱μ-E 曲线Fig.3 XAFS spectrumμ-E curve of Fe3+ (Fe2 O3)and Fe2+(FeCl2)
2.1 XANES分析
由于价态和结构环境的不同,同一种元素在单质、不同化合物中有不同的吸收边,即所谓的吸收边化学漂移[8].对于同一种金属元素,价态越高,吸收边值越大.因此,吸收边值可以用于判断过渡金属的价态.这种基于吸收边值的特征来判断待测物价态的方法称之为“指纹”技术.因为煤灰中可能含有Fe3+和Fe2+,如果含有的高价离子多,则吸收边向高能量方向移动.表2给出了高灰熔点和低灰熔点煤灰样吸收曲线峰位.经过计算处理后,比较结果可以发现,2种样品中Fe的吸收边值处于标准样Fe3+(Fe2O3)和 Fe2+(FeCl2)的吸收边值之间,说明煤灰样中铁原子以Fe3+和Fe2+2种形式同时存在.

表2 高灰熔点和低灰熔点煤灰样吸收曲线峰位Tab.2 Peak position of absorption curves for ash samples with high and low fusion point e V
2.1.1 高灰熔点煤灰样
图4给出了A样品K边XANES图谱.观察图4(a)中不同样品归一化后的K边XANES图谱可知,早在软化温度之前,Fe元素的变化就已经开始.随着温度的升高,A样品中Fe的吸收边向左移动,呈现靠近Fe2+吸收边的趋势,且边前峰(图4(a)左上角)的强度降低,可以判定煤灰样中的Fe3+参与了反应,被还原生成了Fe2+.加热到1 300℃后,样品吸收曲线变化不再明显,可见样品到1 300℃时Fe的元素形态和化学环境的变化已经完成.
一般认为边前峰对应原子1s-3d的跃迁.理论上,在X射线吸收谱中,1s-3d的跃迁是禁止的,因此一般看不到边前峰.但在非中心对称结构(如畸形八面体和四面体)中存在终态d-p轨道杂化,使得d轨道部分具有p轨道的特征,因此能够看到边前峰.杂化程度越高,边前峰强度越高.四面体结构具有最大的d-p杂化程度,表现在XANES图谱中为最高的边前峰[8].由图4(a)左上角的边前峰局部放大图可知,随着温度的升高,Fe的K边边前峰强度降低,可见Fe2+占据八面体配位点,Fe的配位结构由四面体向八面体转变.
对归一化的吸收曲线求一次导数得到图4(b).A样品在900~1 300℃温度段内,尤其是900~1 100℃,X射线的吸收情况变化剧烈,可以判断Fe的元素形态和化学环境的变化大都发生在900~1 300℃的温度段.Zhang等[4]指出熔融过程中煤灰中铁系矿物质(如方铁矿(FeO)、贵榴石(3FeO·Al2O3·3SiO3)、铁橄榄石(FeO·SiO2)等)的产生在1 273~1 373 K的温度段内会影响煤灰在还原性条件下的熔融特性.以上结合物的形态同时验证了本次实验中900~1 300℃温度段内生成Fe2+的结论.
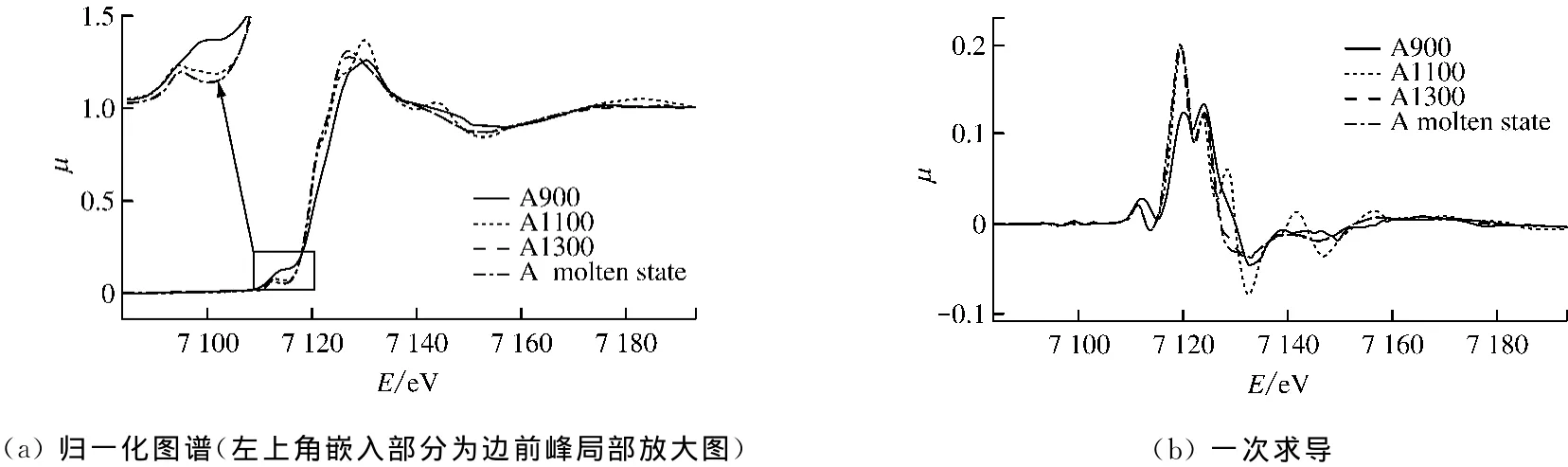
图4 A样品K边XANES图谱Fig.4 K-edge XANES spectrum of sample A
2.1.2 低灰熔点煤灰样
图5给出了B样品K边XANES图谱.观察图5(a)中不同加热温度下B样品归一化后的K边XANES图谱可知,与A样品类似,B样品中Fe的吸收边向左移动,呈现靠近Fe2+吸收边的趋势,可以判定灰样中的Fe3+参与了反应,被还原生成了Fe2+.但不同的是,随着温度的升高,B样品的K边XANES图谱边前峰的强度有一个先上升后下降的过程,而且在样品完全熔融时吸收主峰发生了分裂现象.
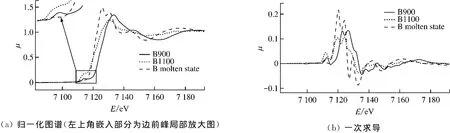
图5 B样品K边XANES图谱Fig.5 K-edge XANES spectrum of sample B
边前峰的强度先上升后下降的现象表明,Fe原子轨道的d-p杂化程度先增大后减小,即Fe原子首先占据四面体中心,然后才向八面体转变.由于煤灰的熔融是复杂的物理化学过程,低灰熔点煤灰样熔融过程中Fe的配位不是由简单的四面体向八面体转变.
主峰出现分裂现象[9],即出现精细结构.对K边吸收曲线来讲,产生这种现象是由于配位体分布的不对称破坏了p轨道系的简并,引起能量不同的p轨道跃迁,从而产生不同的峰.综上说明低灰熔点煤灰样熔融时,Fe的配位变化是Fe2+首先占据四面体空位中心,然后再由四面体配位向八面体配位转变.
2.2 EXAFS分析
对标准样和实验样品的μ-E曲线进行处理,去除原子背景,将能量转换为波数,通过傅里叶转变,将K空间转换成R空间,得到径向结构函数(RSF),如图6和图7所示,其中R为配位层与原子核之间的距离,x(R)表示各配位层散射波的调制叠加.各吸收主峰代表中心Fe原子周围的近邻配位原子吸收入射X射线而发出的光电子散射,表征Fe的不同配位层,因而可以通过拟合个别峰的方法求得对应配位层的结构参数[10].
图6(a)和图7(a)中标准样Fe2O3的第1个峰位于1.4Å处,属于Fe—O配位峰;第2个峰位于2.5Å处,属于Fe—Fe配位峰.标准样FeCl2的第1个峰位于1.6Å处,属于Fe—Cl配位峰;第2个峰位于2.2Å处,属于Fe—Fe配位峰.对于Fe的第1层配位结构,A煤灰样随着温度的升高,Fe—O配位峰位置与标准样Fe2O3大致一致,说明在高灰熔点煤灰样的熔融过程中,Fe的Fe—O配位结构未发生明显的变化.对于B煤灰样,Fe的Fe—O配位在完全熔融前没有发生明显的变化,当B煤灰样完全熔融时,配位峰偏移到1.7Å的位置,说明完全熔融态的低灰熔点煤灰样中Fe的配位结构发生了转变.Fe的第2层配位Fe—Fe在煤灰样中未表现出明显的规律.
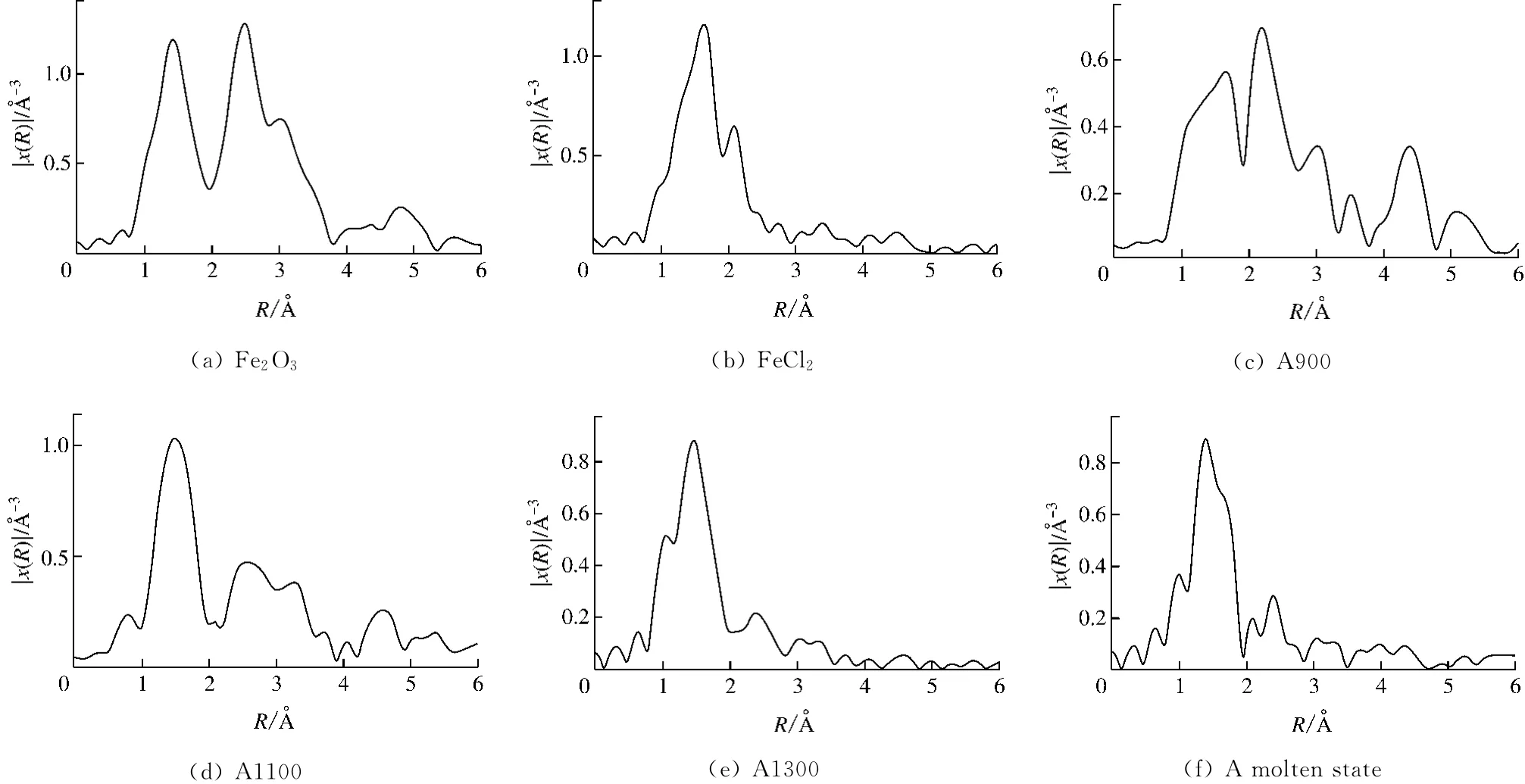
图6 标准样及A煤灰样的RSF谱Fig.6 RSF spectrum of standard sample and sample A
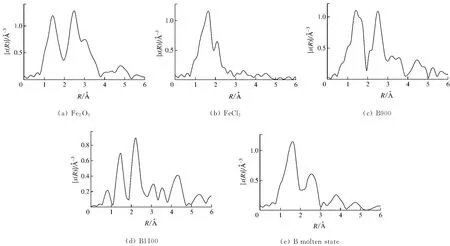
图7 标准样及B煤灰样的RSF谱Fig.7 RSF spectrum of standard sample and sample B
3 结 论
(1)在还原性气氛下煤灰样熔融过程中,Fe的价态形式由Fe3+向Fe2+转变.高灰熔点煤灰样熔融过程中Fe的价态变化大都发生在900~1 300℃的温度段,熔融过程中,Fe由四面体配位向八面体配位转变.但低灰熔点煤灰样熔融时,一部分Fe会首先占据四面体空位中心,然后再向八面体配位转变.Fe元素的这些形态变化在熔融过程开始之前就已经发生.
(2)低灰熔点煤灰样在熔融过程中,第1层配位Fe—O结构发生了明显变化,而且只发生在完全熔融时刻;而高灰熔点煤灰样则没有明显观察到此配位结构的变化.
[1]杨建国,邓芙蓉,赵虹,等.煤灰熔融过程中的矿物演变及其对灰熔点的影响[J].中国电机工程学报,2006,26(17):122-126.YANG Jianguo,DENG Furong,ZHAO Hong,et al.Mineral conversion of coal-ash in fusing process and the influence to ash fusion point[J].Proceedings of the CSEE,2006,26(17):122-126.
[2]陈冬林,杜洋,邹婵,等.不同碱酸比煤灰在刚玉质耐火材料上的结渣特性[J].动力工程学报,2013,33(4):256-260.CHEN Donglin,DU Yang,ZOU Chan,et al.Slagging characteristics of coal ash with different alkali/acid ratios on corundum-based refractory liner[J].Journal of Chinese Society of Power Engineering,2013,33(4):256-260.
[3]张德祥,龙永华,高晋生.煤灰中矿物的化学组成与灰熔融性的关系[J].华东理工大学学报,2003,29(6):590-594.ZHANG Dexiang,LONG Yonghua,GAO Jinsheng.Relationship between the coal ash fusibility and its chemical composition[J].Journal of East China University of Science and Technology,2003,29(6):590-594.
[4]ZHANG Zhongxiao,WU Xiaojiang,ZHOU Tuo,et al.The effect of iron-bearing mineral behavior on ash deposition during coal combustion[J].Processing of the Combustion Institute,2010,33(2):2853-2861.
[5]KONINGSBERGER D C,PRINS R.X-ray absorption:principles,applications,techniques of EXAFS,SESAFS and XANES[M].New York:John Wily,1988.
[6]OYANAGI H,SAKAMOTO K,SHIODA R,et al.Ge overlayers on Si(001)studied by surface-extended X-ray-absorption fine structure[J].Physical Review B,1995,52(2):824-829.
[7]LUO Jinyong,MENG Ming,ZHA Yuqing,et al.A comparative study of Pt/Ba/Al2O3and Pt/Fe-Ba/Al2O3NSR catalysts:New insights into the interaction of Pt-Ba and the function of Fe[J].Applied Ca-talysis B:Environmental,2008,78(1):38-52.
[8]杨修春,刘维学.X射线吸收精细结构谱在材料科学中的应用[J].功能材料,2005,63(8):1146-1150.YANG Xiuchun,LIU Weixue.Application of X-ray absorption fine structure spectroscopy in materials science[J].Journal of Functional Materials,2005,63(8):1146-1150.
[9]罗金勇,孟明.高分散CuO/CeO2-Al2O3催化剂中铜物种的精细结构表征[J].无机化学学报,2006,22(5):861-866.LUO Jinyong,MENG Ming.Characterization on the fine structure of the copper species in the highly dispersed CuO/CeO2-Al2O3catalysts[J].Chinese Journal of Inorganic Chemistry,2006,22(5):861-866.
[10]BAKER L L.Fe K-edge XAFS spectra of phyllosilicates of varying crystallinity[J].Physics and Chemistry of Minerals,2012,39(8):675-684.
猜你喜欢
赣南师范大学学报(2022年6期)2022-12-12
数学物理学报(2021年4期)2021-08-30
煤化工(2021年3期)2021-07-14
数学物理学报(2021年2期)2021-06-09
陶瓷学报(2021年1期)2021-04-13
上海建材(2019年1期)2019-04-25
化工进展(2015年6期)2015-11-13
华东理工大学学报(自然科学版)(2015年1期)2015-11-07
橡胶工业(2015年5期)2015-08-29
中国新技术新产品(2014年5期)2014-07-30
