
铬系催化乙烯配位聚合/齐聚分子模拟研究进展
2014-03-28 06:07程瑞华何雪莲刘柏平
化学反应工程与工艺 2014年3期
刘 振,程瑞华,何雪莲,田 洲,刘柏平
(华东理工大学化学工程联合国家重点实验室,上海200237)
上世纪50年代初,Phillips石油公司Hogan等[1]发明了硅胶负载型铬系乙烯聚合催化剂。经过半个多世纪的发展,每年用Phillips铬系催化剂生产的高密度聚乙烯产品占世界总产量的一半左右[2]。与Ziegler-Natta和茂金属催化剂相比,Phillips铬系催化剂在引发乙烯聚合反应中不需要添加任何烷基金属助催化剂,这也使得其活性中心上聚乙烯的链引发机理备受关注[3]。尽管Phillips铬系催化剂在工业中得到非常广泛的应用,但是有关其活性中心结构、铬的活性价态以及引发乙烯聚合的反应机理等诸多方面至今仍存在争议[2-7]。传统的实验光谱表征方法虽然可以获得与活性中心相关的一些结构和价态信息,但这不足以对当前的一些机理提案提供足够的支持。随着计算机硬件水平的迅速提高和计算化学理论的发展,目前计算机分子模拟已经成为一种新兴的研究方法,被广泛应用于化学和生物学的各个研究领域。在Phillips铬系催化剂的相关研究中,通过采用计算机分子模拟技术与传统实验相结合的方法,可以获得单一方法难以实现的机理认识[3]。
在乙烯聚合反应过程中,往往需要添加一种线性α烯烃作为共聚单体来提高聚烯烃产品性能,这也使得以1-己烯为代表的线性α烯烃的工业用量与日剧增[8]。传统的制备工艺主要是通过乙烯非选择性齐聚和后续分离来获得不同种类的线性α烯烃,采用这种方式制备线性α烯烃的成本也因此大大增加。近年来,乙烯选择性齐聚制备线性α烯烃(主要是1-己烯和1-辛烯)的技术已成为研究热点[8-10]。其中,Chevron-Phillips公司于2003年首次实现了乙烯选择性三聚制备1-己烯的工业化[11],而乙烯选择性四聚也由Sasol公司成功中试[10],这两种催化体系也都同属于铬系催化剂的范畴。此外,近期有关乙烯选择性三聚、四聚催化剂的报道也主要以铬系催化剂为主[8-13]。本工作将从计算机分子模拟的角度,介绍Phillips铬系催化剂引发乙烯聚合反应机理和铬系催化乙烯选择性三聚的研究进展。
1 Phillips铬系催化剂
Phillips铬系催化剂的组成包括硅胶载体和铬活性组分。一般将铬源(CrO3或Cr(CH3COO)3)负载于硅胶载体表面,通过500~900 ℃的高温焙烧制得Phillips铬系乙烯聚合催化剂。Phillips催化剂经过焙烧后,其铬物种通常以单铬酸酯、双铬酸酯和多铬酸酯的形式负载于硅胶表面,然后与CO2等还原剂或者乙烯单体接触后,六价铬酸酯被还原至低价态(通常为二价),随后表现出高的乙烯聚合活性。由于活化后的Phillips催化剂不需要添加烷基金属助催化剂即可引发乙烯聚合反应,这个特点使得Phillips催化剂铬活性中心上第一个铬碳键和第一根聚乙烯链的形成机理备受关注[3]。
通常认为,Phillips铬系催化剂引发乙烯聚合反应机理有Cossee机理、卡宾机理和金属环状机理。早在1964年,Cossee在研究Ziegler-Natta催化剂乙烯聚合反应机理时提出了著名的Cossee机理[14]。由于Ziegler-Natta催化剂需要经过烷基金属预活化处理,因此,Cossee机理能够很好地解释烯烃配位-插入的聚合过程。而Phillips催化剂铬活性中心并不需要经过烷基金属预活化步骤,因此,Cossee机理难以解释第一个铬碳键形成所需要的额外氢的来源(图1a)。卡宾机理则认为乙烯在铬中心上通过分子内的氢转移进行,即形成了铬碳卡宾双键[15](图1b)。金属环状机理则认为链引发反应通过两分子乙烯同时在铬中心配位,然后经过氧化加成反应形成了铬金属五元环结构[16](图1c)。

图1 典型的乙烯聚合链引发反应机理Fig.1 Typical initiation mechanisms for ethylene polymerization
上述催化机理一般都是根据实验光谱表征结果对可能发生的反应进行演绎推理的提案。由于传统实验手段仍然难以在分子原子水平捕获催化反应中间体,这也使得人们对催化机理的认识长期停滞不前。近年来,计算机分子模拟已被广泛应用于各种化学反应过程研究,特别是以密度泛函理论(DFT)为代表的分子模拟方法,在催化反应机理中的应用十分广泛。Phillips催化剂铬活性中心可以被乙烯单体直接还原,并以低价态(主要是二价)的形式负载于硅胶表面,其活性的高低主要取决于铬中心周围电子环境及其空间自由度的影响。Espelid等[17-20]针对Phillips铬系催化剂活性中心的特点,构建了一系列不同类型的Phillips催化剂铬活性中心团簇模型,其中典型的单Cr(II)活性中心如图2中1a,2a和3a所示。Espelid等的DFT计算结果支持1a是最有可能的Phillips铬系催化剂活性中心模型[17]。随后,Demmelmaier等[21]也通过实验和分子模拟研究,支持具有铬金属六元环构型的1a模型为Phillips催化剂的活性中心结构。Damin等[22]则利用CO和N2作为探针分子,考察了其在1a,2a和3a模型上的吸附振动光谱,发现基于1a模型的计算结果与实验数据吻合较好。随后,模型1a也被广泛应用于Phillips铬系催化剂的相关机理研究中[23-26]。由于Phillips铬系催化剂中的硅胶载体对铬活性中心起着重要作用,为了更好地反映硅胶载体对Phillips铬系催化剂活性中心的影响,Zhong等[27]在考察Phillips催化剂活性中心结构时,构建了硅胶负载型活性中心模型(4a),Guesmi等[28]则构建了包含120个原子的无定形硅胶载体模型(5a)。上述研究结果仍支持铬六元环是最有可能的Phillips催化剂铬活性中心模型。下面将从Phillips铬系催化剂诱导期活性中心转换机理以及Ti改性对Phillips催化剂的影响两个方面,对有关Phillips铬系催化剂机理研究方面的工作进展进行介绍。

图2 Phillips铬系催化剂活性中心模型Fig.2 Molecular models for the chromium active site of the Phillips catalyst
1.1 乙烯聚合诱导期内活性中心转换机理
Phillips铬系催化剂引发乙烯聚合反应诱导期内,普遍认为六价铬首先被乙烯单体还原至低价态。Liu等[29]通过程序升温脱附-质谱实验表征方法对Phillips铬系催化剂诱导期内可能发生的反应进行了研究,发现六价铬在被乙烯单体还原的同时,伴随着副产物甲醛的生成,并且有丙烯、丁烯等产物生成,Liu等针对该实验结果,提出了在诱导期内副产物甲醛在铬中心的配位可能引起乙烯聚合活性中心向乙烯易位活性中心转换。针对这一假设,Zhong等[24]对Phillips铬系催化剂引发乙烯聚合诱导期内的反应进行了分子模拟研究,其构建了三种活性中心模型,如图3所示。其中1a为二价铬乙烯聚合活性中心,模型1b的铬中心配有一分子甲醛,而模型2b的铬中心则同时配有两分子甲醛。

图3 Phillips铬系催化剂甲醛吸附活性中心分子模型以及诱导期内各反应势能图示Fig.3 Molecular models and energy profiles for the reactions over model 1a during the induction period of the Phillips catalyst
以1a模型为例,两分子乙烯可能同时在铬中心配位形成双乙烯-铬配合物53b,然而在高自旋五重态势能面通过协同氧化加成反应形成铬金属五元环物种54b需要克服高达177.4 kJ/mol的能垒,这也极大地阻碍了反应继续进行。该研究的计算结果表明,在铬金属五元环形成的过程中存在一个最低能量交叉点,反应经过5-3CP1从高自旋五重态势能面交叉至低自旋三重态势能面进行,整个过程只需要克服能垒99.2 kJ/mol。随后,从铬金属五元环34b在自旋三重态势能面上向四元环35b转换形成乙烯易位活性中心需要克服高达239.3 kJ/mol的能垒,因此,该步反应难以发生。这也表明在没有甲醛配位的条件下,二价铬活性中心难以形成乙烯易位活性中心。从铬五元环34b到生成1-丁烯的反应是通过两步氢转移进行的,其中,铬金属五元环中的β氢首先向铬中心转移生成36b,随后经过β氢消去生成1-丁烯。在第二步氢转移过程中也存在类似的最低能量交叉点(3-5CP2),使第二步反应的能垒降低了34.3 kJ/mol。而从铬金属七元环39b生成1-己烯则是通过分子内一步氢转移进行的,在1-己烯形成之后,反应经过另外一个最低能量交叉点(3-5CP3),生成了更稳定的五重态1-己烯配合物510b。
表1列出了副产物甲醛分子在铬中心上的吸附对催化剂活性中心影响的计算结果。当铬中心配位一分子甲醛时(模型1b),则第三分子乙烯无法在铬五元环物种上进一步配位,同时,由铬五元环向易位活性中心转换的能垒也降低了43.9 kJ/mol,这表明甲醛分子的吸附有利于铬中心由乙烯聚合中心向乙烯易位中心转换。当铬中心同时吸附两分子甲醛时(模型2b),由于甲醛分子的屏蔽效应阻碍了乙烯分子的配位,使铬中心发生失活。钟蕾等[30]进一步采用氟改性活性中心模型对上述反应进行了计算,其结果列于表1。针对氟改性的计算结果表明,当没有甲醛分子配位时,聚合活性中心向易位活性中心转换、乙烯二聚和三聚反应步骤的能垒都有不同程度的提高,而进行环增长反应的能垒则降低了22.5 kJ/mol,这表明氟改性有利于乙烯聚合反应的进行。当配位一分子甲醛时,氟改性更有利于聚合中心向易位中心的转换以及1-丁烯的形成。

表1 Phillips铬系催化剂诱导期内各反应步骤的活化能以及甲醛分子配位和氟改性的影响Table 1 Energy barriers of the proposed reaction pathways during the induction period of the Phillips catalyst with considering the effects of formaldehyde adsorption and fluorination
1.2 Ti改性催化剂乙烯聚合机理
Phillips铬系催化剂在工业中可以生产多种分子量分布的聚烯烃产品,其活性中心会受到铬中心周围电子环境和空间立体效应的显著影响。通常情况下,采用Ti改性的硅胶载体可以提高催化剂的活性以及降低聚合物的分子量[2]。最近,Cheng等[23]通过实验与计算机分子模拟方法,对Ti改性的Phillips铬系催化剂乙烯聚合行为进行了研究。该研究构建了六种代表不同Ti含量的铬活性中心模型,其中包括3种二价铬模型和3种六价铬模型,如图4所示。其中,模型1a和3c表示没有经过Ti改性的活性中心;1c和4c表示Ti/Cr为1:1的活性中心;2c和5c表示Ti/Cr为2:1的活性中心。
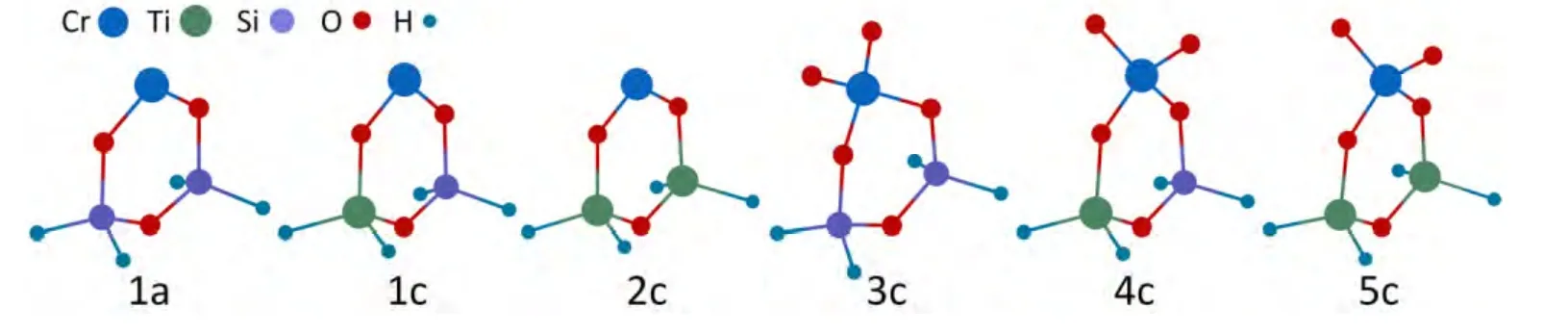
图4 Ti改性Phillips铬系催化剂活性中心模型Fig.4 Molecular models for the active sites of Ti-modified Phillips catalyst
一般而言,乙烯聚合速率主要由聚合物链增长的快慢决定,而聚合物分子量的高低则由链转移与链增长的相对速率决定。如图5所示,该研究通过设计不同Ti/Cr活性中心模型,分别计算了乙烯单体插入聚合物链以及增长中的聚合物链通过链转移进行终止反应的过程。

图5 典型的乙烯聚合过程链增长和链转移步骤Fig.5 Schematic representation of the typical chain propagation and chain transfer reactions for ethylene polymerization
针对图4中三种还原态二价铬活性中心模型,其上进行链增长和链转移反应的能垒如表2所示。当Ti/Cr依次增加时,乙烯插入聚合物链的能垒分别为80.3,77.0和74.1 kJ/mol。这表明Ti改性有利于乙烯聚合速率的提高,从而也提高了催化剂的活性。聚合物的分子量和分子量分布通常由乙烯链增长和链转移之间的相对速率决定。采用Ti改性活性中心模型的计算结果表明,Ti改性可以降低乙烯链增长和链转移步骤的反应能垒,但是Ti改性使得链转移步骤的能垒降低更多,因此,提高了链转移与链增长之间的相对速率,这与实验中观测到Ti改性Phillips铬系催化剂更倾向于生产低分子量的聚乙烯产品的结果一致[2]。

表2 Ti改性Phillips铬系催化剂乙烯聚合链引发、链增长和链转移步骤的反应能垒Table 2 Energy barriers for chain initiation, chain propagation (1, 2-insertion), and chain transfer (β-H elimination to monomer) reactions over various models of the Phillips catalyst
在乙烯聚合过程中,通过加入共聚单体可以提高聚烯烃产品的性能。表2中的数据同时也表明,Ti改性降低了共聚单体插入步骤的能垒,同时降低了通过β氢向共聚单体发生链转移的能垒,而且后者降低的程度更大。这说明Ti改性Phillips催化剂生产的聚烯烃产品中共聚单体的含量将降低,同时插入的共聚单体更有利于β氢消去反应。因此,Ti改性的Phillips催化剂生产的聚合物低分子量部分的短支链含量也更低。
在传统的Ziegler-Natta催化剂和Phillips铬系催化剂上,烯烃通常以1,2-插入方式进行链增长反应。该研究通过计算共聚单体1-丁烯和1-己烯分别以1,2-插入和2,1-插入方式进行链增长的能垒,结果如表3所示。

表3 Ti改性Phillips铬系催化剂共聚单体插入步骤的反应能垒Table 3 Energy barriers through regiospecific insertions of comonomers over various models of the Phillips catalyst
在所有三种催化剂活性中心模型上,1,2-插入的能垒均低于2,1-插入步骤的能垒,这也证实了反应过程中1,2-插入是主要的α烯烃插入方式。通过对比不同Ti含量活性中心模型的计算数据可以发现,Ti组分的引入,使得2,1-插入过程的能垒降低更多,表明Ti改性的Phillips铬系催化剂加强了烯烃的2,1-插入方式,这个结论也与McDaniel的实验推断一致[2]。
2 铬系乙烯选择性齐聚催化剂
Phillips铬系催化剂在工业中生产着不同牌号和不同密度的聚烯烃产品。为了改善产品性能和调节产品密度,往往需要在乙烯聚合过程中添加一定比例的共聚单体,最为常见的有1-丁烯、1-己烯和1-辛烯等线性α烯烃。由于当前1-丁烯的生产工艺比较成熟,产品相对比较便宜,因此,1-丁烯作为共聚单体在聚烯烃的生产中用量最大。但是,1-己烯和1-辛烯作为共聚单体可以进一步提高聚乙烯产品的性能,这也使得以1-己烯为代表的线性α烯烃的工业用量剧增[2]。在传统的工业生产中,1-己烯通常采用乙烯非选择性齐聚的方式进行生产。但这种方式需要对乙烯齐聚产物进行后续分离,不仅生产工艺较为繁琐,也增加了1-己烯的工业生产成本。目前,Shell,BP Amoco和Chevron Phillips等传统的乙烯齐聚技术和Sasol的Fischer-Tropsch的1-己烯生产技术通常只能得到一系列的线性α烯烃的混合物[11]。
1967年,Manyik等[31]在申请乙烯连续聚合过程工艺的相关专利时,提到Cr(III) 2-ethylhexanoate(Cr(III) 2-EH)体系在加入部分水解的三异丁基铝(PIBAO)后,可以催化乙烯聚合反应,其中1-己烯是齐聚副产物的主要组分。Manyik等[32]于1977年首次报道了Cr(III) 2-EH/PIBAO体系可以催化乙烯选择性三聚,产物中主要成分为1-己烯。经过近30年的研究,Chevron Phillips公司在2003年成功地实现了2,5-dimethylpyrrole/Cr(III)/triethylaluminium体系催化乙烯选择性三聚的工业化生产[11]。该催化体系对乙烯的选择性高达90%以上,其主要的改进就是在反应体系中添加了2,5-二甲基吡咯配体和诸如三乙基铝/一氯二乙基铝等烷基金属助催化剂。
如前所述,乙烯聚合机理常见的有Cossee链增长机理、卡宾机理和金属环状反应机理。由于Cossee机理通常只能生成全分布的线性α烯烃,卡宾机理也只能通过易位反应生成具有奇数和偶数碳的全分布线性α烯烃。而金属环状反应路径则被普遍认为是乙烯选择性齐聚的反应机理。在这个反应过程中,环增长和β-氢消去的竞争是决定生产何种类型α烯烃的关键因素。如图6所示,文献中关于乙烯三聚机理的提案主要包括路径a,路径b和路径c。其中路径a最早由Manyik等[32]提出,反应经过了β氢向单体转移的关键中间体;路径b由Briggs等[33]提出,反应经过了β氢向对位α碳转移生成含有不饱和链的中间物种;而路径c由Deckers等[34]提出,反应经过了β氢向铬中心转移的中间体。

图6 文献中报道的乙烯选择性三聚反应机理Fig.6 Various reaction mechanisms for selective ethylene trimerization proposed in the literature
当前,实验与分子模拟研究普遍支持乙烯选择性三聚遵循金属环状反应机理。Cr(III) 2-EH/PIBAO体系作为首次报道的乙烯三聚反应体系,通过加入dimethoxyethane(DME)组分,反应可以由乙烯聚合向乙烯三聚发生转换。此外,有实验研究报道,Cr-SNS(SNS=RS(CH2)2N(H)(CH2)2SR)乙烯三聚反应体系中可能存在着Cr(I)/Cr(III)和Cr(II)/Cr(IV)两种催化循环。因此,下面将从Cr(III) 2-EH/PIBAO/DME体系的聚合、三聚转换机理以及Cr-SNS体系中铬活性中心价态两个方面对本研究室在乙烯选择性三聚机理研究方面的工作进展进行介绍。
2.1 Cr-DME催化乙烯三聚机理
Manyik等[31,32]首次发现Cr(III) 2-EH/PIBAO乙烯聚合体系的副产物中主要成分为乙烯三聚产物1-己烯。随后,Briggs[33]报道了Cr(III) 2-EH/PIBAO/DME催化剂体系对乙烯的选择性可以达到74%。因此,DME组分可以使Cr(III) 2-EH/PIBAO体系由乙烯聚合催化体系向乙烯三聚催化体系转换。Qi等[35]通过DFT计算对该转化机理进行了研究,考察了DME对乙烯选择性三聚反应的影响。如图7所示,在乙烯三聚过程中,三聚产物1-己烯是铬七元环通过β氢转移生成的,该步骤记为T,而铬七元环的进一步增长则可以认为反应继续向乙烯聚合方向进行,该步反应记为P。

图7 乙烯选择性三聚关键步骤竞争反应Fig.7 Schematic representation of the competing reactions for the key step of selective ethylene trimerization
该研究设计了一系列活性中心模型,包括三组阳离子活性中心模型和两组中性模型。表4列出了基于上述模型分别进行β氢消去和环增长步骤所对应的能垒。对于阳离子模型,在没有DME配位时,反应更倾向于环增长反应,也就是说1d,2d和3d阳离子中心更有可能是乙烯聚合活性中心。DME在铬中心上的配位,增加了铬周围的空间位阻,因此,提高了乙烯分子进一步插入进行环增长反应步骤的能垒,这也使得反应更有利于发生β氢转移,从而生成乙烯三聚产物1-己烯,这与实验报道一致。但是,在中性活性中心模型4d和5d上,在不添加DME组分的情况下,铬活性中心便有利于β氢消去生成1-己烯的反应,因此,中性模型可能不是Cr(III) 2-EH/PIBAO/DME催化体系的乙烯三聚活性中心。

表4 DME对乙烯选择性三聚生成1-己烯和进一步环增长成九元环步骤的反应能垒的影响Table 4 Energy barriers of 9-membered ring formation (P) and 1-hexene liberation via agostic-assisted β-H transfer (T) over different models with considering the effect of DME coordination
2.2 Cr-SNS催化乙烯三聚机理
如前所述,研究者普遍认为乙烯选择性三聚通过金属环状反应机理进行。但是乙烯三聚催化体系中铬中心的活性价态依然存在争议。在2003年,McGuinness等[36]报道了SNS-CrCl3体系在甲基铝氧烷(MAO)存在条件下表现出极高的乙烯三聚选择性和活性。McGuinness等提议在MAO还原过程中,SNS配体可能发生了去质子化反应。然而,Jabri等[37]发现去质子化的SNS-Cr(II)催化体系未能表现出乙烯三聚活性。Yang等[38]针对Cr-SNS体系,构建了一组未去质子化的模型1e和2e及其对应的去质子化模型3e和4e(图8),对该体系催化乙烯选择性三聚反应机理进行了计算机分子模拟研究,在研究过程中主要考察了关键中间体铬金属五元环和铬金属七元环物种的形成步骤,如图8中右图所示。

图8 Cr-SNS乙烯三聚体系的分子模型及关键反应步骤Fig.8 Molecular models for Cr-SNS catalyst system and a schematic representation of the key steps for ethylene trimerization
该研究发现在Cr-SNS催化的乙烯选择性三聚过程中,模型1e,3e和4e上乙烯三聚反应过程中发生了电子自旋交叉现象,因此,表5列出了电子自旋交叉对铬五元环和七元环形成步骤能垒的影响。显然,在不考虑电子自旋交叉时,去质子化的一价模型3e最有可能形成铬金属七元环,进而通过β氢消去生成1-己烯。在考虑电子自旋交叉后,尽管模型3e上形成铬金属五元环步骤的能垒显著降低,然而在形成七元环步骤的能垒却高达169.0 kJ/mol,这极大地阻碍了1-己烯的形成。事实上,在考虑电子自旋交叉之后,未去质子化的模型1e上发生乙烯三聚的两个关键能垒均为127 kJ/mol左右。综合考虑两个关键步骤的能垒,未去质子化模型1e更有利于1-己烯的生成。这表明,Cr-SNS体系中未去质子化的Cr(I)-SNS最有可能是乙烯三聚活性中心,三聚反应依从Cr(I)/Cr(III)催化循环进行。

表5 乙烯选择性三聚过程生成铬五元环和铬七元环步骤的反应能垒Table5 Energy barriers of formation of metallacyclopentane (MCP) and metallacycloheptane (MCH) over different molecular models without/with considering the spin surface crossing
3 展 望
计算机分子模拟与实验相结合可以获得单一实验手段无法达到的认识深度。比如,计算机分子模拟可以对Phillips铬系催化剂诱导期内可能发生的化学反应在分子原子水平上进行解析,可以直接解明Ti改性对Phillips铬系催化剂的影响方式,可以确定铬系催化乙烯三聚体系中活性物种的价态,可以探寻铬系催化反应过程中可能发生的电子自旋交叉现象。然而,上述的计算机分子模拟结果要与实验紧密结合,根据分子模拟提出的电子自旋交叉现象还有待于实验的进一步验证。此外,当前针对Phillips催化剂的分子模拟还基本停留在通过建立小型团簇模型来研究铬活性中心催化行为的阶段。由于工业Phillips催化剂铬中心是负载于无定形硅胶载体表面,并且受到硅胶载体孔隙率和表面积等多种因素的影响,因此,在随后的计算机分子模拟中需要建立更为真实的硅胶负载型活性中心模型。随着当代计算机硬件水平的提升,以及计算化学理论和方法的发展,这部分工作的开展已变成可能。
分子模拟与实验密切相关。一方面,实验结果可以对分子模拟方法的选择、模型的建立起到一定的指导作用,反过来,计算机分子模拟的结果也能够使研究者更为深入地理解化学反应机理,从而可以有目的地改进实验,最终实现由传统试错型研究到现代瞄准中的型研究的转变。可以预见,在不久的将来,计算机分子模拟会深入化学化工研究的各个方向,并且会发挥越来越重要的作用。不断提升的计算机硬件水平可以帮助研究更为复杂的物质体系,通过在计算机上开展催化剂的分子设计和高通量筛选,可以大大缩短催化剂开发周期和降低催化剂开发成本,更有效地进行催化剂开发。总之,烯烃聚合领域的发展需要该领域的科学工作者进行密切合作,通过多学科交叉和融合,采用多种有效的实验方法和理论模拟相结合的手段,共同努力向前推进工业催化领域理论和应用研究的发展。
[1] Hogan J P, Banks R L. Polymers and production thereof: US, 2825721[P]. 1958-03-04.
[2] McDaniel M P. A review of the Phillips supported chromium catalyst and its commercial use for ethylene polymerization[J]. Advances in Catalysis, 2010, 53: 123-606.
[3] Groppo E, Lamberti C, Bordiga S, et al. The structure of active centers and the ethylene polymerization mechanism on the Cr/SiO2catalyst: a frontier for the characterization methods[J]. Chemical Reviews, 2005, 105(1): 115-183.
[4] McDaniel M P. Supported chromium catalysts for ethylene polymerization[J]. Advances in Catalysis, 1985, 33: 47-98.
[5] McDaniel M P. Some reflections on the current state of Cr-based polymerization catalysts[J]. MRS Bulletin, 2013, 38(3): 234-238.
[6] Cheng Ruihua, Liu Zhen, Zhong Lei, et al. Phillips Cr/silica catalyst for ethylene polymerization[J]. Advances in Polymer Science, 2013,257: 135-202.
[7] Liu Zhen, He Xuelian, Cheng Ruihua, et al. Chromium catalysts for ethylene polymerization and oligomerization[J]. Advances in Chemical Engineering, 2014, 44: 127-191.
[8] McGuinness D S. Olefin oligomerization via metallacycles: dimerization, trimerization, tetramerization, and beyond[J]. Chemical Reviews, 2011, 111(3): 2321-2341.
[9] Agapie T. Selective ethylene oligomerization: recent advances in chromium catalysis and mechanistic investigations[J]. Coordination Chemistry Reviews, 2011, 255(7-8): 861-880.
[10] van Leeuwen P W N M, Clément N D, Tschan M J L. New processes for the selective production of 1-octene[J]. Coordination Chemistry Reviews, 2011, 255(13-14): 1499-1517.
[11] Dixon J T, Green M J, Hess F M, et al. Advances in selective ethylene trimerization: a critical overview[J]. Journal of Organometallic Chemistry, 2004, 689(23): 3641-3668.
[12] 宋宪凤, 毕四勇, 毛国梁, 等. 乙烯三聚和四聚催化剂的研究进展[J]. 石油化工, 2008, 37(8): 858-862.Song Xianfeng, Bi Siyong, Mao Guoliang, et al. Advances in catalysts for trimerization and tetramerization of ethylene[J]. Petrochemical Technology, 2008, 37(8): 858-862.
[13] 宋宪凤, 宁英男, 姜 涛, 等. 乙烯选择性四聚制1-辛烯催化剂及工艺技术研究进展[J]. 化工进展, 2008, 27(5): 693-696.Song Xianfeng, Ning Yingnan, Jiang Tao, et al. Advance in research of the catalyst and technology for ethylene tetramerization toward 1-octene[J]. Chemical Industry and Engineering Progress, 2008, 27(5): 693-696.
[14] Cossee P. Ziegler-Natta catalysis I: mechanism of polymerization of alpha-olefins with Ziegler-Natta catalysts[J]. Journal of Catalysis,1964, 3(1): 80-88.
[15] Ghiotti G, Garrone E, Zecchina A. IR investigation of polymerization centers of the Phillips catalyst[J]. Journal of Molecular Catalysis,1988, 46(1-3): 61-77.
[16] Groppo E, Lamberti C, Bordiga S, et al. In situ FTIR spectroscopy of key intermediates in the first stages of ethylene polymerization on the Cr/SiO2Phillips catalyst: Solving the puzzle of the initiation mechanism?[J]. Journal of Catalysis, 2006, 240(2): 172-181.
[17] Espelid O, Borve K J. Theoretical models of ethylene polymerization over a mononuclear chromium(II)/silica site[J]. Journal of Catalysis,2000, 195(1): 125-139.
[18] Espelid O, Borve K J. Theoretical analysis of CO adsorption on the reduced Cr/silica system[J]. Journal of Catalysis, 2002, 205(1):177-190.
[19] Espelid O, Borve K J. Molecular-level insight into Cr/silica Phillips-type catalysts: polymerization-active mononuclear chromium sites[J].Journal of Catalysis, 2002, 205(2): 366-374.
[20] Espelid O, Borve K J. Molecular-level insight into Cr/silica Phillips-Type catalysts: polymerization-active dinuclear chromium sites[J].Journal of Catalysis, 2002, 206(2): 331-338.
[21] Demmelmaier C A, White R E, van Bokhoven J A, et al. Evidence for a chromasiloxane ring size effect in Phillips(Cr/SiO2)polymerization catalysts[J]. Journal of Catalysis, 2009, 262(1): 44-56.
[22] Damin A, Vitillo J G, Ricchiardi G, et al. Modeling CO and N2adsorption at Cr surface species of Phillips catalyst by hybrid density functionals: effect of Hartree-Fock exchange percentage[J]. The Journal of Physical Chemistry A, 2009, 113(52): 14261-14269.
[23] Cheng Ruihua, Xu Chen, Liu Zhen, et al. High-resolution spectroscopy (XPS,1H MAS solid-state NMR) and DFT investigations into Ti-modified Phillips CrOx/SiO2catalysts[J]. Journal of Catalysis, 2010, 273(2): 103-115.
[24] Zhong Lei, Liu Zhen, Cheng Ruihua, et al. Active site transformation during the induction period of ethylene polymerization over the Phillips CrOx/SiO2catalyst[J]. ChemCatChem, 2012, 4: 872-881.
[25] Liu Zhen, Cheng Ruihua, He Xuelian, et al. DFT functional benchmarking on the energy splitting of chromium spin states and mechanistic study of acetylene cyclotrimerization over the Phillips Cr(II)/silica catalyst[J]. The Journal of Physical Chemistry A, 2012,116(28): 7538-7549.
[26] Liu Zhen, Cheng Ruihua, He Xuelian, et al. Reactivity and regioselectivity of methylacetylene cyclotrimerization over the Phillips Cr/silica catalyst: a DFT study[J]. ACS Catalysis, 2013, 3: 1172-1183.
[27] Zhong Lei, Lee Mingyung, Liu Zhen, et al. Spectroscopic and structural characterization of Cr(II)/SiO2active site precursors in model Phillips polymerization catalysts[J]. Journal of Catalysis, 2012, 293: 1-12.
[28] Guesmi H, Tielens F. Chromium oxide species supported on silica: a representative periodic DFT model[J]. The Journal of Physical Chemistry C, 2012, 116(1): 994-1001.
[29] Liu B, Nakatani H, Terano M. New aspects of the induction period of ethylene polymerization using Phillips CrOx/SiO2catalyst probed by XPS, TPD and EPMA[J]. Journal of Molecular Catalysis A: Chemical, 2002, 184(1-2): 387-398.
[30] 钟 蕾, 刘 振, 程瑞华, 等. 载体硅胶表面氟改性对Phillips铬系催化剂诱导期内引发乙烯聚合行为的影响[J]. 化工学报, 2013,64(2): 539-546.Zhong Lei, Liu Zhen, Cheng Ruihua, et al. Effect of fluorination of silica support on initiation of ethylene polymerization during induction period over Phillips catalyst[J]. CIESC Journal, 2013, 64(2): 539-546.
[31] Manyik R M, Walker W E, Wilson T P. Continuous processes for the production of ethylene polymers and catalysts suitable there for field: US, 3300458[P]. 1967-01-24.
[32] Manyik R M, Walker W E, Wilson T P. Soluble chromium-based catalyst for ethylene trimerization and polymerization[J]. Journal of Catalysis, 1977, 47(2): 197-209.
[33] Briggs J R. The selective trimerization of ethylene to hex-1-ene[J]. Journal of the Chemical Society, Chemical Communications, 1989, 11:674-675.
[34] Deckers P J W, Hessen B, Teuben J H. Switching a catalyst system from ethylene polymerization to ethylene trimerization with a hemilabile ancillary ligand[J]. Angewandte Chemie, International Edition, 2001, 40(13): 2516-2519.
[35] Qi Yuan, Dong Qi, Zhong Lei, et al. Role of 1,2-dimethoxyethane in the transformation from ethylene polymerization to trimerization using chromium tris(2-ethylhexanoate)-based catalyst system: a DFT study[J]. Organometallics, 2010, 29(7): 1588-1602.
[36] McGuinness D S, Wasserscheid P, Keim W, et al. First Cr(III)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene[J]. Journal of the American Chemical Society, 2003, 125(18) :5272-5273.
[37] Jabri A, Temple C, Crewdson P, et al. Role of the metal oxidation state in the SNS-Cr catalyst for ethylene trimerization: isolation of diand trivalent cationic intermediates[J]. Journal of the American Chemical Society, 2006, 128(28): 9238-9247.
[38] Yang Yun, Liu Zhen, Zhong Lei, et al. Spin surface crossing between chromium(I)/sextet and chromium(III)/quartet without deprotonation in SNS-Cr mediated ethylene trimerization[J]. Organometallics, 2011, 30(19): 5297-5302.
猜你喜欢
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
化工管理(2020年26期)2020-10-09
环境与生活(2016年11期)2016-12-02
合成化学(2015年2期)2016-01-17
山西大同大学学报(自然科学版)(2015年1期)2015-01-22
中国造纸(2014年1期)2014-03-01
无机化学学报(2014年8期)2014-02-28
中国有色金属学报(2011年4期)2011-11-08
