
Population Dynamics Following the Last Glacial Maximum in Two Sympatric Lizards in Northern China
2014-03-25 02:10,*
Asian Herpetological Research 2014年4期
,*
1Jiangsu Key Laboratory for Biodiversity and Biotechnology, College of Life Sciences, Nanjing Normal University, Nanjing 210023, Jiangsu, China
2Hangzhou Key Laboratory for Animal Adaptation and Evolution, School of Life Sciences, Hangzhou Normal University, Hangzhou 310036, Zhejiang, China
Population Dynamics Following the Last Glacial Maximum in Two Sympatric Lizards in Northern China
Yanfu QU1, Qun ZHAO1,2, Hongliang LU2and Xiang JI1*
1Jiangsu Key Laboratory for Biodiversity and Biotechnology, College of Life Sciences, Nanjing Normal University, Nanjing 210023, Jiangsu, China
2Hangzhou Key Laboratory for Animal Adaptation and Evolution, School of Life Sciences, Hangzhou Normal University, Hangzhou 310036, Zhejiang, China
Phylogeographic studies of Eremias lizards (Lacertidae) in East Asia have been limited, and the impact of major climatic events on their population dynamics remains poorly known. This study aimed to investigate population histories and refugia during the Last Glacial Maximum of two sympatric Eremias lizards (E. argus and E. brenchleyi) inhabiting northern China. We sequenced partial mitochondrial DNA from the ND4 gene for 128 individuals of E. argus from nine localities, and 46 individuals of E. brenchleyi from fi ve localities. Forty-four ND4 haplotypes were determined from E. argus samples, and 33 from E. brenchleyi samples. Population expansion events began about 0.0044 Ma in E. argus, and 0.031 Ma in E. brenchleyi. The demographic history of E. brenchleyi indicates a long-lasting population decline since the most recent common ancestor, while that of E. argus indicates a continuous population growth. Among-population structure was signifi cant in both species, and there were multiple refugia across their range. Intermittent gene flow occurred among expanded populations across multiple refugia during warmer phases of the glacial period, and this may explain why the effective population size has remained relatively stable in E. brenchleyi and grown in E. argus.
Lacertidae, Eremias lizards, mitochondrial DNA, historical demography, multiple refugia, Last Glacial Maximum
1. Introduction
Historic climatic change (e.g., climatic oscillations during the Pleistocene Epoch) affected the historical demography of extant species (Hewitt, 2000, 2004). Glacial and interglacial periods consisted of a series of alternating warming periods of limited duration (interstadials) and short-term cooling periods (stadials) (Martrat et al., 2004). The effective population size of a species may decrease during the glacial period and increase during the interglacial period. However, if there were multiplerefugia within its range, the effective population size of the species might remain stable or grow during the glacial period due to intermittent gene flow between refugia during warmer periods (interstadial) (Li et al., 2009; Ruiz-González et al., 2013). Climatic changes during Pleistocene cycles have played a key role in shaping genetic diversity of species and left genetic signatures in present populations (Hewitt, 1996, 1999, 2000, 2004; Avise, 2000) that can help identify glacial refugia, post-glacial migration routes and population dynamics (Newton et al., 1999; Petit et al., 2005; Li et al., 2009; Tian et al., 2009; Ruiz-González et al., 2013). Relative to birds and mammals, reptiles are less able to disperse and are therefore more sensitive to climatic fl uctuations, making them more suitable biogeographical indicators (Camargo et al., 2010; Malhotra et al., 2011).
Lacertid lizards of the genus Eremias are widely distributed in Southeast Europe, West and Central Asia, and Mongolia, northern China, Korea and Russia (regions of Lake Baikal) in East Asia. Of some 30 extant species, eight can be found in China, six of which (E. argus, E. arguta, E. brenchleyi, E. grammica, E. velox and E. vermiculata) are oviparous, and two (E. multiocellata and E. przewalskii) are viviparous (Zhao, 1999). The ancestor of Eremias lizards has been hypothesized to originate from Europe and dispersed northwards to Central Asia through Africa during the late Miocene 8−10 million years ago (Ma) (Fu, 1998; Arnold et al., 2007; Mayer and Pavlicev, 2007; Hipsley et al., 2009). According to this hypothesis, Eremias lizards found in East Asia should spread from west (Central Asia) to east, not following westward, southward or northward adaptive radiation of species found in other parts of the world (Forcina et al., 2012; Mori et al., 2013; Zink et al., 2013). During the Last Glacial Maximum (LGM), multiple glacial refugia were available to East Asian species throughout their ranges (Qian and Ricklefs, 2000; Li et al., 2009; Tian et al., 2009; Bai et al., 2010) primarily due to a relatively mild Pleistocene climate in that region (Weaver et al., 1998; Ju et al., 2007). However, as phylogeographic studies of Eremias lizards in East Asia have been limited, the impact of major climatic events on their population dynamics remains poorly known.
Here, we study two oviparous Eremias lizards (E. argus and E. brenchleyi) sympatric throughout the distribution of E. brenchleyi in the central and northern parts of China (Chen, 1991). The two species can coexist primarily because they use different microhabitats (E. argus mainly inhabits plains and hills, while E. brenchleyi inhabits hills only) not because they differ greatly along the resource axes of diet and time (Zhao, 1999). Both species are relatively common and have more intact population genetic structure than other species or populations that have become threatened or endangered in China (Elderkin et al., 2007; Lin et al., 2010; Zhao et al., 2011), thus providing ideal model systems in which to investigate population histories, glacial refugia and postglacial migration routes. Here, we used partial mtDNA sequences from the gene ND4 to address three questions regarding population dynamics following LGM in these two species. (1) Did they survive in multiple refugia across their species range or in a single refuge during the LGM? (2) What was the pattern of recolonization routes? (3) How did effective population size vary during the LGM?
2. Materials and Methods
2.1 SamplingWe sequenced the mitochondrial ND4 gene from 128 individuals of E. argus from nine sites (Figure 1 A, Table 1) and 46 individuals of E. brenchleyi from fi ve sites (Figure 1 B, Table 1). The most distal 2−3 mm of the tail tip from each lizard was stored in 95% ethanol. Lizards were released at their site of capture immediately after tissue sampling. The infl uence of tail breaks at the distal portion of the tail often is not signifi cant in lizard species with caudal autotomy, including Eremias lizards (Lin and Ji, 2005; Lin et al., 2006; Zhao et al., 2008; Sun et al., 2009; Ding et al., 2012). Voucher specimens were deposited at Nanjing Normal University and assigned voucher numbers identified by locality-haplotype numbers. The sampling code, geographic information and genetic analysis are given in Appendix A, Appendix B and Table 1. We used all sequences from this study and three sequences from two lacertid lizards (Lacerta viridis viridis: NC_008328; Timon lepidus: DQ902321 and DQ902322) as ingroups, and two sequences (DQ150379 and DQ150376) from Psammodromus algirus (Hipsley et al., 2009) as outgroups for the phylogenetic analysis based on ND4 haplotypes.
2.2 Molecular dataGenomic DNA was isolated from tail samples using standard phenol-chloroform extraction methods (Palumbi, 1996). Fragments of complete ND4 gene from the mitochondrial genome were amplified with the primers ND4 (5′-CAC CTA TGA CTA CCA AAA GCT CAT GTA GAA GC-3′) and leu (5′-CAT TAC TTT TAC TTG GAT TTG CAC CA-3′) (Busack et al., 2005). PCRs were performed in 100 μl volumes, using a hot start method; PCR cycling parameters were 94 °C for 7 min; 40 cycles of 94 °C for 40 s, 46 °C for 30 s, 72 °C for 1 min; and a fi nal elongation step of 72 °C for 7 min. PCR products were then purified with a spin column containing Sepacry S-400 (Amersham Bioscience AB, Uppsala, Sweden) and sequenced using both forward and reverse primers on an ABI-PRISMTM310 Genetic Analyzer (Applied Biosystems Information, USA). Sequences were edited and aligned manually using BIOEDIT version 7.0.9.0 (Hall, 1999), and were translated to amino acids with Mega 6.0 to verify if a functional mitochondrial DNA sequence was obtained and that nuclear pseudogenes were not amplified (Tamura et al., 2013). All sequences were deposited in GenBank under accession numbers JN561171−JN561247.

Figure 1 Map of northern China showing localities where individual E. argus (A) and E. brenchleyi (B) were collected. See text and Table 1 for sample locality abbreviations. Numbers in parentheses are sample sizes. Small circles represent sampling localities, and pie charts represent haplotype group frequencies within each locality. The blue, dotted lines outline the species range in China.
2.3 Phylogenetic analysisPhyolgenetic analysis were carried out by Bayesian inference (BI) using MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003) and MrModeltest 2.3 (Nylander, 2004). The best-fit model (GTR + I) for 1st codon position, (HKY + G) for 2nd codon position, and (GTR + G) for 3rd codon position were selected by Akaike Information Criterion (AIC) in MrModeltest 2.3. Four Markov Chains Monte Carlo (MCMC) samples were run for 5 × 106iterations. Two independent runs were carried out to allow additional confirmation of the convergence of MCMC runs. Trees were sampled
every 500 iterations, providing 2 × 104samples from the two runs. The first 4% of the iterations were discarded as burn-in, leaving 1.96 × 104samples to estimate the consensus tree and the Bayesian posterior probabilities. Haplotypes with lineages were evaluated using medianjoining networks (MJNs) in NETWORK 4.5.1.0 (Bandelt et al., 1999).
2.4 Divergence timesWe used Bayesian MCMC method to evaluated divergence times and evolutionary rates. ForE. argus, we set the mean of the prior distribution for the time separating the ingroup root from the present (rttm) to 45.4 Ma estimated based on the age (44.1−46.7 Ma with a mean of 45.4 Ma; Appendix A, node 5 (Hipsley et al., 2009) of the split between Eremias and Timon. The standard deviation of the prior distribution for the time separating the ingroup root from the present (rttmsd) was set at one-half of rttm. The mean and standard deviation of prior distribution for rate at root node (rtrate and rtratesd) were set to 0.008 2 substitutions per site per 1 Ma, with the rtrate value calculated by dividing the median distance between the ingroup root and the tips by rttm; the mean and standard deviation of prior for brownian motion constant nu (brownmean and brownsd) both were set to 0.022, so that rttm × brownmean = 1, following recommendations accompanying the software. All remaining parameters were set to default values. In this study, the fossil calibration point was defined to the most recent common ancestor of E. argus according to the Middle Pleistocene (0.438−0.548 Ma) found in Chinese fossil layers (Li et al., 2004) (node N3 in Figure 2). The Bayesian analysis was run for 5 × 106generations, with a sample frequency of 100 after a burn-in of 5 × 104generations.
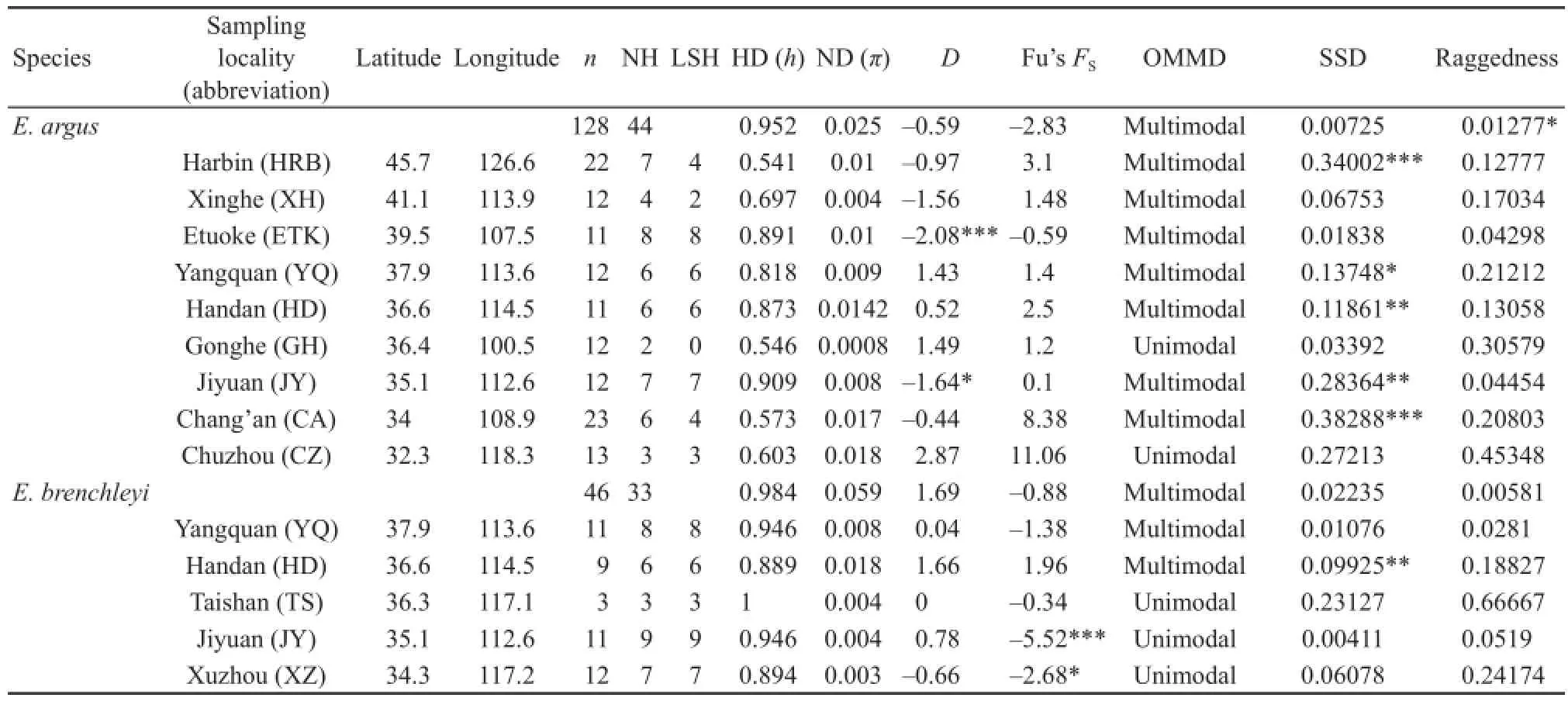
Table 1 Descriptive statistics by sampling locality for the two lizards studied. NH: number of haplotypes; LSH: locality-specifi c haplotypes; HD: haplotype diversity; ND: nucleotide diversity; D: Tajima’s D; Fu’s FS: statistics of Fu’s FStest (1997); OMMD: observed modality of mismatch distribution; SSD: the sum of square deviation between the observed and simulated mismatch distributions; Raggedness: raggedness index.
To estimate intraspecies divergence time of E. brenchleyi, we set rttm to posterior distribution of the divergence time of Eremias and Timon, which came from the above results of E. argus intraspecies divergence-time estimation rttmsd was set to equal rttm. The rtrate value was calculated by dividing the median distance between the ingroup root and the tips by rttm and the rtratesd value was set to equal rtrate. We set all the remaining parameters to default values. We used two calibration points in these estimates (N15 and N16 in Figure 3). The N16 calibration point was based on the fossil record of ancestor of E. argus from the Middle Pleistocene (0.438−0.548 Ma) (Li et al., 2004). The lower bound for the age of node N16 was 0.438 Ma, and the upper bound for the age of node N16 was 0.548 Ma. N15 was not a fossil calibration but a secondary calibration point from the above results of the 95% confi dence interval of E. argus intraspecies divergence-time estimation. The Bayesian analysis was run for 5 × 106generations, with a sample frequency of 100 after a burn-in of 5 × 104generations.
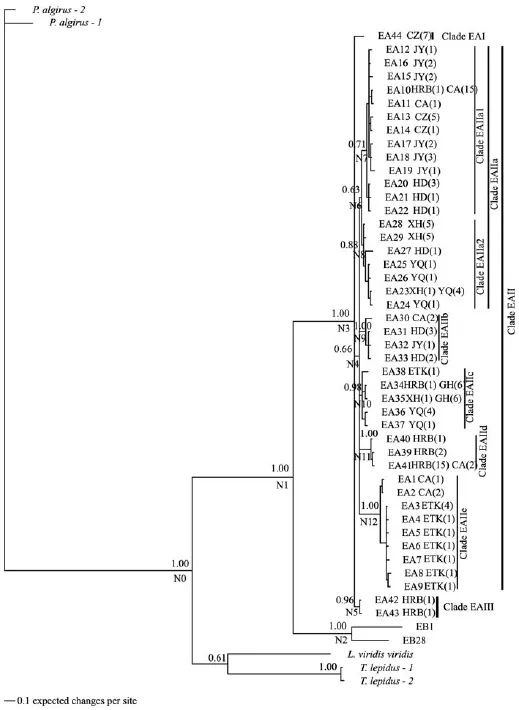
Figure 2 The partitioned Bayesian phylogenetic tree based on ND4 haplotypes for E. argus. Nodes N0 to N12 denote internal nodes of interest. Numbers above the branches are Bayesian posterior probabilities. Taxa are haplotypes; all haplotype designations are listed in Appendix C, followed by the sampling localities and numbers of individuals from each locality having that haplotype [e.g., JY(1)].
2.5 Population genetic analysesWe followed Zhao et al. (2011) to calculate haplotype diversity (h), mean nucleotide diversity (π), mismatch distributions, Fu’s Fs and Tajima’s D for each sampling site and for each species as a whole (1000 replicates). Nucleotide diversity and haplotype diversity were tested for a significant correlation with latitude using linear regression. We performed mismatch-distribution tests for goodness-offi t against a null model of sudden population expansion calculated using SSD between observed and expected data, and Harpending’s raggedness Index (Fu, 1997; Excoffi er et al., 2005). To further estimate past population dynamics through time, we constructed BSP implemented in BEAST 1.6.1 (Drummond and Rambaut, 2007) with a relaxed molecular clock method to depict the change of the two species’ effective population size since the time to the most recent common ancestor (TMRCA) of the sampled ND4 haplotypes respectively. For each BSP, the model of molecular evolution was assessed for the ND4 sequence by AIC in MrModeltest 2.3 (Nylander, 2004). We selected the best-fi t models (GTR + G) and (GTR + I + G) for E. argus and E. brenchleyi, respectively. The only one calibration point based on the fossil record from the Middle Pleistocene (0.438−0.548 Ma) (Li et al., 2004) was assigned to the most recent common ancestor (MRCA) of E. argus and for this species we assumed a normal prior distribution centered at 0.493 Ma with a standard deviation of 0.028 Ma for the MRCA. For E. brenchleyi, the evolutionary rate of ND4 was set to the ucld.mean of BSP of E. argus above because no fossil
calibration points are available. MCMC sampling was run for 3 × 107generations, with a sample frequency of 100 after a burn-in of 3 × 105generations. The effective sample size of each parameter was measured and demographic plots for the two species were visualized in Tracer version 1.5 (Rambaut and Drummond, 2007). We also made the diagram of oxygen isotopic (δ18O) record during the past 0.78 Ma as proxy for global ice distribution for each species (Imbrie et al., 1984).
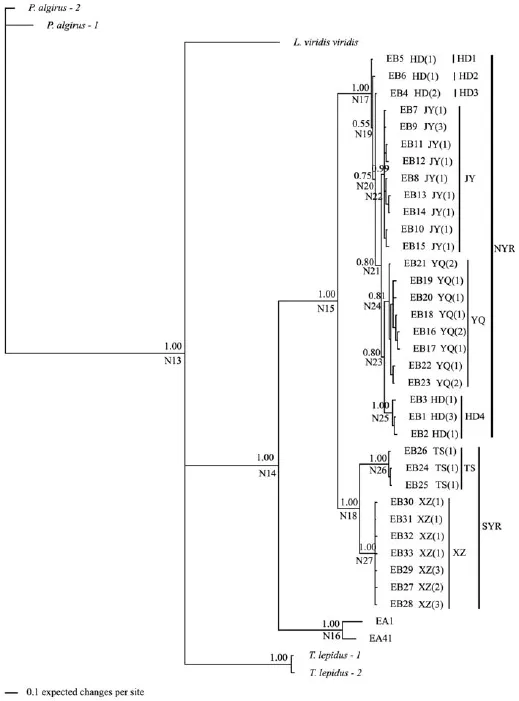
Figure 3 The partitioned Bayesian phylogenetic tree based on ND4 haplotypes for E. brenchleyi. Nodes from N13 to N27 denote internal nodes of interest. Numbers above the branches are Bayesian posterior probabilities. Taxa are haplotypes; all haplotype designations are listed in Appendix C, followed by the sampling localities and numbers of individuals from each locality having that haplotype [e.g., JY(1)].
We computed the pairwise ΦSTvalue for each combination of populations using the Kimura twoparameter model of nucleotide substitution (Kimura, 1981), which takes into account haplotype frequencies and the genetic distance between haplotypes in ARLEQUIN. The significance of ΦSTvalues was evaluated using 1000 permutations of the data and set at P = 0.05. We performed a Mantel test to compare the pairwise population structure estimates to a matrix of geographical distance. We used ARLEQUIN also to perform a two-level hierarchical analysis of molecular variance (AMOVA) (Excoffier et al., 1992) to test for structure among populations: within populations, and
among populations (ΦST).
One additional procedure was used to characterize major patterns of genetic structure across the ranges sampled for both species. We applied the SAMOVA procedure (Dupanloup et al., 2002) to identify groups of collection localities that were maximally differentiated based on ND4, and to infer genetic barriers between these groups. We used SAMOVA 1.0 to perform analyses based on 100 simulated annealing steps and examined the number of groups (k) that maximized genetic differentiation among groups (ΦCT). The analysis was run for k = 2 to k = n (n = 8 for E. argus; n = 4 for E. brenchleyi) and the signifi cance level of fi xation indices was evaluated by 1000 permutations.
3. Results
3.1 Matrilineal patternsWe obtained 44 E. argus haplotypes (Appendix A) and 33 E. brenchleyi haplotypes (Appendix B). The ratio of haplotypes relative to the total number of individuals sampled was 0.34 for E. argus and 0.71 for E. brenchleyi. Neither mean nucleotide diversity (F1,12= 0.69, P = 0.42) nor the number of haplotypes (F1,12= 0.96, P = 0.35) per sampling location differed between the two species (Table 1). The number of haplotypes, the number of unique haplotypes, haplotype diversity and nucleotide diversity for single sampling localities were not correlated with latitude in either species (all P > 0.26). All E. argus haplotypes formed a single well-supported clade, which could be subdivided into three clades (EA I, EA II and EA III) although the phylogenetic relationships among these clades were not fully resolved (PP < 0.50) (Figure 2). Clade EA I included a single haplotype (EA44) from the Chuzhou (CZ) population. Clade EA II was poorly supported (PP = 0.66) and further subdivided into fi ve clades: one poorly supported clade (EA IIa, PP = 0.63) and four well-supported clades (EA IIb, PP = 1.00; EA IIc, PP = 0.98; EA IId, PP = 1.00; EA IIe, PP = 1.00). These five clades were mostly admixed in many local populations and had no geographical specificity. Clade EA IIa was further subdivided into two poorly supported clades (EA IIa1, PP = 0.71; EA IIa2, PP = 0.88) that were mostly admixed in the Handan (HD) population and had no geographical specificity. Clade EA III included two haplotypes from the Harbin (HRB) population.
All E. brenchleyi haplotypes formed a single wellsupported clade, which could be subdivided into two well-supported clades [north (NYR) and south (SYR) of the Yellow River, both PP = 1.00] (Figure 3). The NYR clade included all haplotypes from the Yangquan (YQ), HD and Jiyuan (JY) populations in NYR, and the SYR clade included all haplotypes from the Xuzhou (XZ) and Taishan (TS) populations in SYR. The division of the two clades coincided with the Yellow River (Figure 1). There was a clear population-genetic structure within both clades. The NYR clade could be subdivided into three major clades (HD4, PP = 1.00; JY, PP = 0.99; YQ, PP = 0.81) and three minor clades (HD1, HD2 and HD3) of which each included a single haplotype from the HD population east of the Taihang Mts (36°−40°N, 112°−115°E). The HD4 clade included haplotypes all from the HD population; the JY clade included all haplotypes from the JY population also in east of the Taihang Mts; the YQ clade included all haplotypes from the YQ population west of the Taihang Mts. The HD1, HD2, HD3, HD4 and JY clades formed one group including all haplotypes from two local populations (HD and JY) east of the Taihang Mts; the YQ clade formed the other group including all haplotypes from the YQ population west of the Taihang Mts. The geographical division of these two groups coincided with the Taihang Mts (Figure 1). The SYR clade could be subdivided into two clades with strong nodal support (TS clade and XZ clade, both PP = 1.00). The TS clade included all haplotypes from the TS population, while the XZ clade included all haplotypes from the XZ population.
For E. argus, results of the Median-joining network (MJN) suggests little or no association between haplotypes and geography (Figure 4 A, Appendix A). We detected related haplotypes that formed eight groups (EA I, EA IIa1, EA IIa2, EA IIb, EA IIc, EA IId, EA IIe and EA III) in the Bayesian phylogenetic tree (Figure 2, Figure 4 A). Clade EA IIa2 and EA IIc co-occurred in Xinghe (XH) and YQ. Clade EA IIa1 and EA IIb cooccurred in HD, JY and Chang’an (CA). Clade EA IIa1 and EA IId co-occurred in HRB and CA. Clade EA IIa1, EA IIb, EA IId and EA IIe co-occurred in CA. Clade EA IIa1, EA IIc, EA IId and EA III co-occurred in HRB. For E. brenchleyi, however, we detected networks of related haplotypes that were clustered into two distinct areas (SYR and NYR) respectively corresponding to the SYR clade and NYR clade in the Bayesian phylogenetic tree (Figure 3, Figure 4 B, Appendix B). We further detected networks of related SYR haplotypes that were clustered into two distinct areas (TS and XZ), and detected networks of related NYR haplotypes that were clustered into two distinct areas [YQ and (HD1, HD2, HD3, HD4, and JY)]. These four areas [(HD1, HD2, HD3, HD4, and JY), YQ, TS, and XZ] correspond to clades HD1, HD2, HD3, HD4, and JY, clade YQ, clade TS, and clade XZ,
respectively (Figure 3).
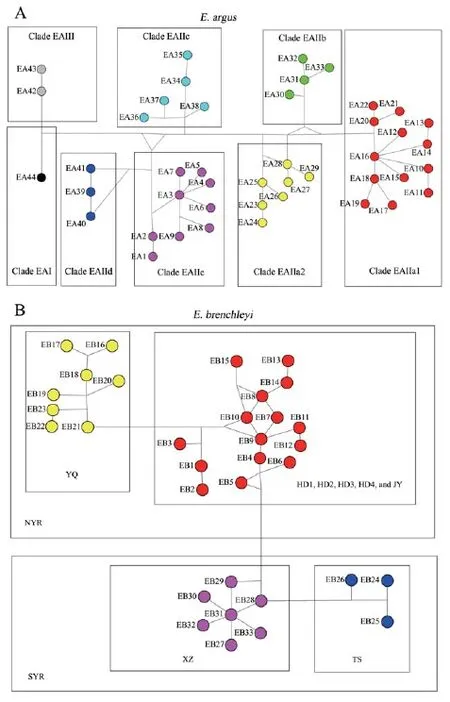
Figure 4 Median-joining networks of mtDNA haplotypes at the ND4 gene in E. argus (A) and E. brenchleyi (B). Numbers adjacent to open circles (network nodes) refer to haplotypes listed in Appendices A and B. Haplotype group color for E. argus is consistent with Figure 1 A, and haplotype group color for E. brenchleyi is consistent with Figure 1 B.
3.2 Divergence timeTable 2 gives posterior distributions of divergence time at each node and 95% credibility intervals based on phylogenies. For E. argus, the most recent common ancestor (TMRCA) of clade EA IIa1 (node N7), EA IIa2 (node N8), EA IIb (node N9), EA IIc (node N10), EA IId (node N11), EA IIe (node N12), and EA III (node N5) were estimated at 0.158 ± 0.063, 0.178 ± 0.064, 0.205 ± 0.087, 0.220 ± 0.082, 0.144 ± 0.082, 0.184 ± 0.082 and 0.100 ± 0.067 Ma, respectively. For E. brenchleyi, the divergence time of the NYR clade from the SYR clade (node N15) was dated to 3.208 ± 0.743 Ma. Within the NYR clade, the divergence time of clades HD1, HD2, HD3, HD4 and JY from clade YQ (node N23) was dated to 0.939 ± 0.418 Ma. Within the SYR clade, the divergence time of the TS clade from the XZ clade (node N18) was dated to 2.095 ± 0.703 Ma. The TMRCA of clades JY and HD (node N17) was estimated at 1.798± 0.615 Ma; the TMRCA of clades YQ (node N24), XZ (node N27) and TS (node N26) were estimated at 0.757 ± 0.369, 0.262 ± 0.264 and 0.404 ± 0.324 Ma, respectively.

Table 2 The posterior distribution of divergence times with 95% confi dence intervals estimated by Bayesian molecular dating. DT: divergence times (Ma); SD: standard deviation; CI: 95% credibility interval.
3.3 Population-genetic analysisFor E. argus, mismatch distributions of the Gonghe (GH) and CZ populations exhibit relatively unimodal patterns with non-signifi cant raggedness values, matching the expected distribution of an expanding population well, and supporting that these two populations have undergone rapid demographic expansion. Mismatch distributions of the remaining seven populations show multimodal distributions. Significant P-values for the sum of squares deviations (SSD), high raggedness indexes, positive Fu’s FSand Tajima’s D values indicate no expansion in the YQ and HD populations, and negative Tajima’s D values suggest recent expansion in the HRB, XH, Etuoke (ETK), JY and CA populations.
For E. brenchleyi, mismatch distributions of the TS, JY and XZ populations exhibit relatively unimodal patterns with on-signifi cant raggedness values, fi tting the expected distribution of an expanding population (non-signifi cant SSD) and supporting that these three populations have undergone rapid demographic expansion. Mismatch distributions of the YQ and HD populations show multimodal distributions. The significant P-value for SSD, high raggedness index, positive Fu’s FSand Tajima’s D values indicate no population expansion in the HD population, and negative Fu’s FSvalue, nonsignifi cant SSD and low raggedness index indicate recent population expansion in the YQ population.
For all populations of E. argus, the multimodal mismatch distribution, non-significant P-value for SSD, low but significant raggedness index and negative Fu’s FSand Tajima’s D values indicate these populations as a whole have possibly undergone recent expansion. The Bayesian skyline plot of E. argus suggested that population size remained relatively stable or grew slowly through time during both cold (positive δ18O values) and warm periods (negative δ18O values) (Figure 5). Increase was followed by a decline starting at about 0.0496 Ma, and then decline was followed by an increase starting at 0.004 39 Ma.
For all populations of E. brenchleyi, negative Fu’s FSvalues indicate populations as a whole have possibly undergone recent expansion. The mismatch distribution showed multimodal, and the P-value for SSD and the raggedness index with low-value both were nonsignificant. To construct Bayesian skyline plots (BSP) of E. brenchleyi, the rate of evolution of ND4 was set to 0.064 5 substitutions per site per Ma (from the ucld. mean of BSP of E. argus). The Bayesian skyline plot of E. brenchleyi implied that population size remained relatively stable or descend slowly through time during both cold (positive δ18O values) and warm periods (negative δ18O values) (Figure 6). Decline was followed by an increase starting at about 0.030 6 Ma. The TMRCA of E. argus and E. brenchleyi were estimated about 0.493 Ma (with the 95% credibility interval ranging from 0.438 to 0.548 Ma) and 1.514 Ma (with the 95% credibility interval ranging from 0.922 to 2.248 Ma), respectively.
Results of AMOVA indicated the presence of geographic genetic structure within both species, concordant with results of each BSP above. For E. argus, a two-level AMOVA to test for structure among populations detected high genetic structure (ΦST= 0.594, P < 0.001). A small portion of the total genetic structure exists within populations, and the majority was among populations (ΦST= 0.594, P < 0.001). For E. brenchleyi, a two-level AMOVA to test for structure among populations
detected relatively higher genetic structure (ΦST= 0.908, P < 0.001). A small portion of the total genetic structure exists within populations, and the majority was among populations (ΦST= 0.905, P < 0.001).
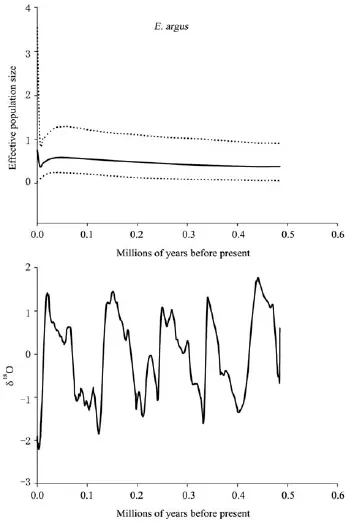
Figure 5 Pleistocene ice distribution (bottom) and Bayesian skyline plot (BSP) (top) representing the historical demographic trend of mitochondrial lineages of E. argus. The solid line represents the mean of the effective population size, while dotted lines represent the 95% HPD limits. The δ18O curve is used as proxy for global ice distribution (data from the SPECMAP project: http://www.ngdc. noaa.gov/mgg/geology/specmap.html). Negative values represent less ice; positive values represent more extensive ice coverage.
For E. argus, pairwise area estimates of ΦSTranged from 0.149 to 0.868. All of the 36 pairwise ΦSTvalues were statistically significant (Table 3). Results of the Mantel test between ΦSTand geographical distance showed a weak relationship between population structure and distance (R2= 0.18, P = 0.096). For E. brenchleyi, pairwise area estimates of ΦSTranged from 0.525 to 0.972. All of the 10 pairwise ΦSTvalues were signifi cant (Table 4). The results of the Mantel test between ΦSTand geographical distance showed a significant relationship between population structure and distance (R2= 0.43, P = 0.007). Thus, only E. brenchleyi shows conclusive evidence of isolation by distance.
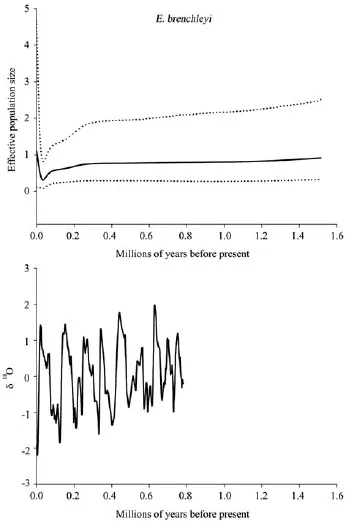
Figure 6 Pleistocene ice distribution (bottom) and Bayesian skyline plot (top) representing the historical demographic trend of mitochondrial lineages of E. brenchleyi. The solid line represents the mean of the effective population size, while dotted lines represent the 95% HPD limits. The oxygen isotopic curve (δ18O) is plotted as proxy for global ice distribution (data from the SPECMAP project: http://www.ngdc.noaa.gov/mgg/geology/specmap.html). Negative values represent less extensive ice coverage; positive values represent more extensive ice coverage.
Table 5 shows results of SAMOVA. For E. argus group structure was maximized at k = 7 (ΦCT= 0.504). The maximum among-group structure was: JY and CA vs. HRB vs. XH and YQ vs. GH vs. ETK vs. HD vs. CZ. For E. brenchleyi, there were distinct groups of genetically defi ned sampling areas, with ΦCTranging from 0.489 to 0.672 when the program was instructed to identify k = 2 through k = 4. Partitions of sampling areas were identifi ed that suggested SYR vs. NYR groups (partitions: YQ, HD and JY vs. TS and XZ; ΦCT= 0.708) at k = 2, and this grouping pattern was concordant with the division of the Yellow River and Bayesian phylogenetic tree (Figure 3). Group structure was maximized at k = 3 (ΦCT= 0.808). However, partitions of sampling areas suggest YQ vs. HDand JY vs. TS vs. XZ [respectively corresponding to clade YQ, (clades HD1, HD2, HD3, HD4 and JY), clade TS and clade XZ in the Bayesian phylogenetic tree (Figure 3)] at k = 4, with only a minimal reduction in group structure (ΦCT= 0.769).
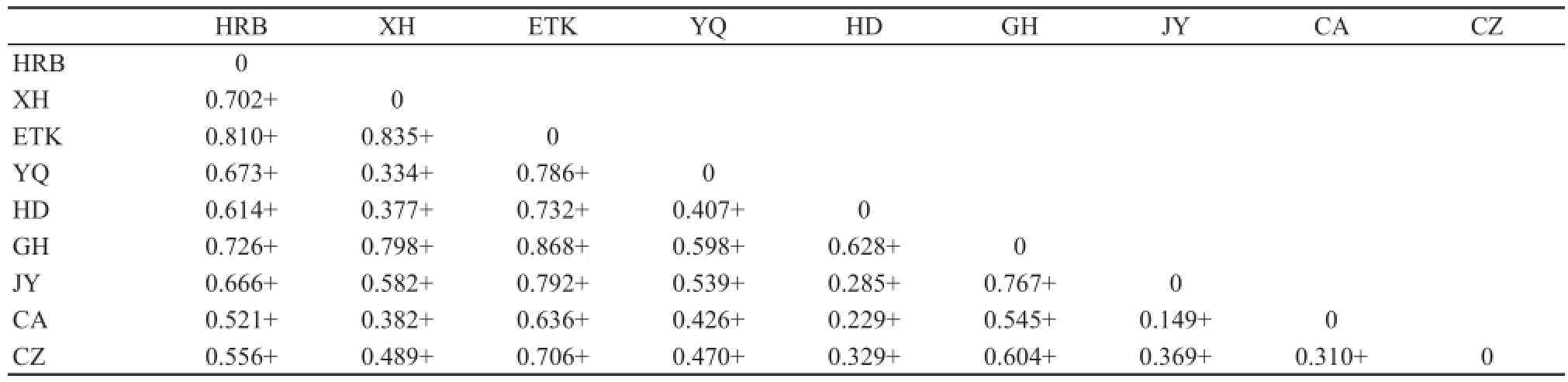
Table 3 Eremias argus pairwise estimates of genetic structure (ΦST), + represents signifi cance at P < 0.05.
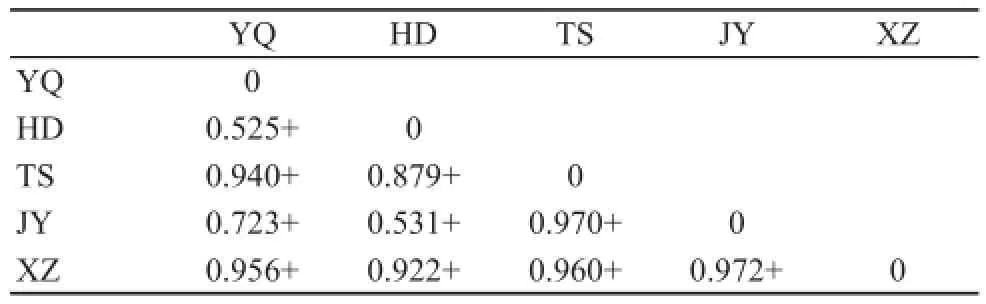
Table 4 Eremias brenchleyi pairwise estimates of genetic structure (ΦST), + represents signifi cance at P < 0.05.
4. Discussion
4.1 Phylogeographical patternsBayesian analysis indicated that E. argus was composed of eight matrilines and had no geographical specificity. Unlike E. argus, there were two reciprocally well-supported lineages (NYR and SYR) within E. brenchleyi, and their geographical separation occurred at the Yellow River (Figure 1, Figure 3). Results of MJN analysis revealed geographic genetic structuring in the two species, coinciding with results of the Bayesian analysis. There are no evident signs of isolation in E. argus, but the Yellow River and Taihang Mts may have acted as important barriers to gene fl ow in E. brenchleyi. SAMOVA analysis of E. brenchleyi indicated that overall haplotypes were clustered into four distinct areas, corresponding to the four phylogeographical units (Avise, 2000) and and concordant with the Bayesian and MJN analyses.
The Yellow River formed during the initial stage of the early Pleistocene (about 1.6 Ma) (Li et al., 1996; Zhang et al., 2003), and the divergence time between the two clades in E. brenchleyi was estimated at 3.208 ± 0.743 Ma (with the 95% credibility interval ranging from 1.648 to 4.309 Ma). Accordingly, we conclude that E. brenchleyi may have been separated by the Yellow River into two lineages (NYR and SYR). This conclusion is consistent with that drawn previously using cyt b gene (Zhao et al., 2011). Within clade NYR, the divergence time of the (HD1+HD2+HD3+HD4+JY) group from the YQ group was dated to 0.939 ± 0.418 Ma (with the 95% credibility interval ranging from 0.283−1.884 Ma). Therefore, the NYR lineage may have been separated by the Taihang Mts into two groups (Figure 1, Figure 3) because Taihang Mts was uplifted to 1100−1500 m during the Pleistocene-Holocene (Wu et al., 1999). The SYR lineage may have been separated into two groups (TS and CZ), although we did not detect the mountain or river with which the geographical division of the clades TS and CZ coincided. The mantel test revealed that E. brenchleyi had more strongly restricted gene fl ow among populations than did E. argus. We suppose that genetic divergence results primarily from the lack of gene flow between the two regions (TS and CZ), probably because they are far enough from each other to result in isolation by distance. Dispersal ability is greater in E. argus than in E. brenchleyi (Zhao et al., 2011), so the Yellow River may be less likely to serve as a barrier for E. argus to disperse between SYR and NYR during Yellow River zerofl ow, and the Taihang Mts are also less likely to act as a boundary between the lineages of E. argus. This conclusion is consistent with that drawn for E. argus based on cyt b gene data (Zhao et al., 2011).
AMOVA analysis indicated that among-population structure was high in E. argus and even higher in E. brenchleyi, consistent with many studies showing that geographic genetic structuring is strong for mtDNA variation in lizards (Clark et al., 1999; Stenson and Thorpe, 2003; Jin et al., 2008; Urquhart et al., 2009). The range of genetic structure is narrower in E. argus (from 0.149 to 0.868) than in E. brenchleyi (from 0.525 to 0.972). Eremias brenchleyi had more signifi cant geographical structuring of haplotype diversity than E. argus.
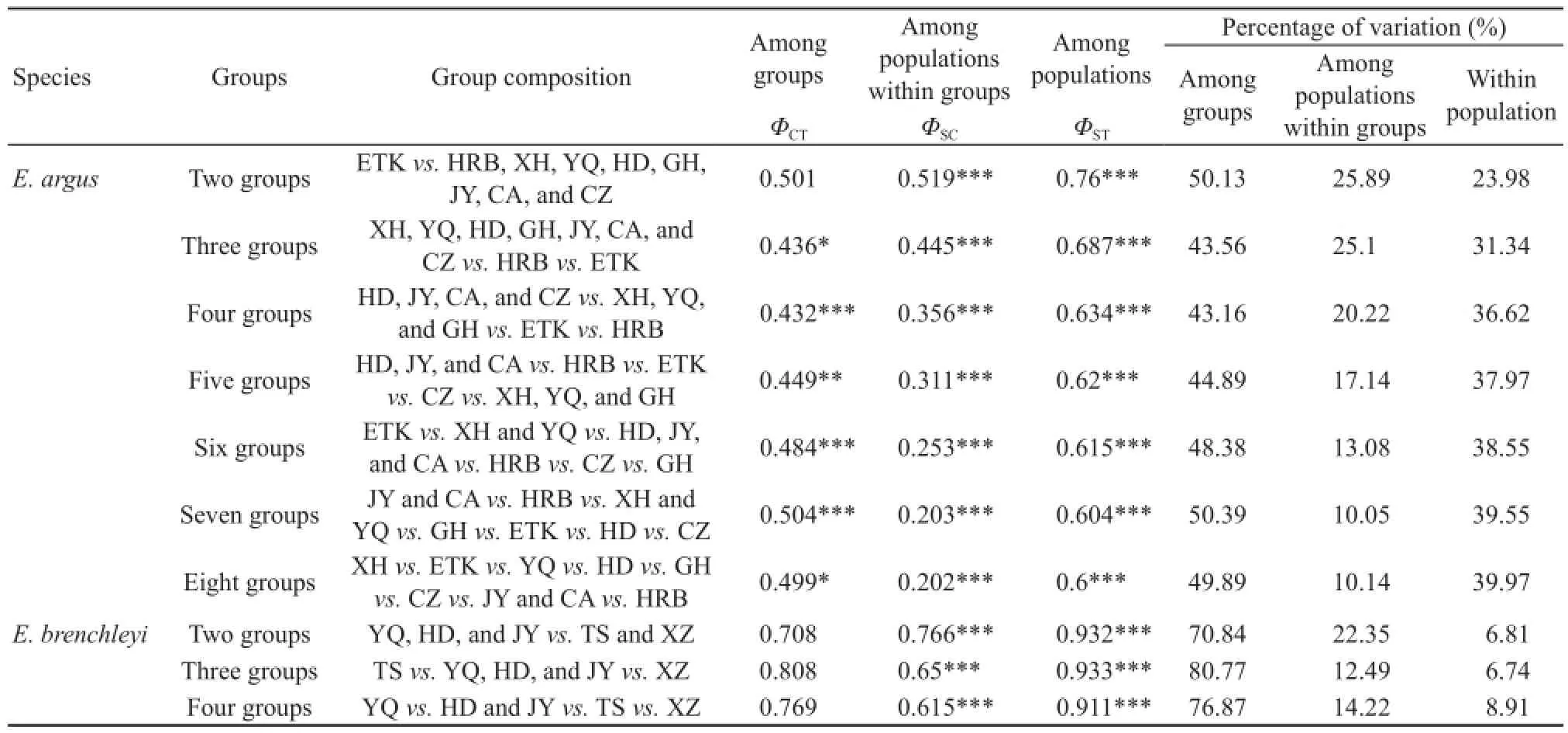
Table 5 Result of SAMOVA using mitochondrial ND4 sequences. The best number of groups has the largest ΦCTvalue
4.2 Genetic diversity and multiple refugiaOur data show that neither in E. argus nor E. brenchleyi are diversity indices correlated with latitude and that genetic diversity is almost homogeneous across the range of each species. These findings are consistent with cyt b data reported previously for the two species (Zhao et al., 2011) but not with phylogeographic studies of widespread terrestrial species in North America and Europe, where they expanded from southern refugia at the end of the Pleistocene, thus exhibiting more geographically structured genetic diversity in the south (Hewitt, 1996, 1999, 2000). It may be the case that multiple refugia were maintained across the range of each species during the LGM, with neither species expanding from a single refuge.
Bayesian analysis shows that the haplotype clade of E. argus is composed of six major clades (EAIIa1, EAIIa2, EAIIb, EAIIc, EAIId and EAIIe) with strong nodal support (all PP > 0.7). The TMRCA of these six clades is estimated to be 0.158 ± 0.063, 0.178 ± 0.064, 0.205 ± 0.087, 0.220 ± 0.082, 0.144 ± 0.082 and 0.184 ± 0.082 Ma, respectively. The haplotype clade of E. brenchleyi is composed of fi ve major clades (JY, YQ, HD4, TS and XZ) with strong nodal support (all PP > 0.8). The TMRCA of these fi ve clades is estimated to be about 0.735 ± 0.394, 0.757 ± 0.369, 0.575 ± 0.355, 0.404 ± 0.324 and 0.262 ± 0.264 Ma, respectively. Thus, all major clades for these two lizards formed before the LGM (about 0.06−0.1 Ma) (Tian et al., 2009). Considering that ancestral haplotypes are more likely to be found at refugia (Slatkin , 1996; Parsons et al., 1997; Li et al., 2009), and the TMRCA of these clades, it is possible that six major clades of E. argus and five major clades of E. brenchleyi diagnose six separate glacial refugia in E. argus and fi ve separate glacial refugia in E. brenchleyi during the LGM.
Furthermore, we propose an alternative hypothesis that each species expanded from a single refuge after the LGM. In this case, related haplotypes could be expected to form a star-like network circling the ancestral haplotype (Avise, 2000), and among-population structure could be expected to be low (Hewitt, 2000). In contrast to these expectations, we detected a reticulated rather than starlike network in both species (Figure 4), and found high among-population structure in E. argus (ΦST= 0.594, P <0.001) and even higher among-population structure in E. brenchleyi (ΦST= 0.908, P < 0.001) that probably resulted from genetic drift favouring/fi xing different alleles among multiple independent refugia during the LGM (Tian et al., 2009). Multiple isolated refugia promote genetic differentiation between current populations (Avise, 2000). Our fi ndings support the existence of multiple refugia in both E. argus and E. brenchleyi, and are consistent with the results reported in similar phylogeographic studies (Hewitt, 1996, 2000; Zamudio and Savage, 2003; Li et al., 2009; Tian et al., 2009). Multiple glacial refugia may have been available to East Asian species throughout their entire ranges (Li et al., 2009), probably because East Asia potentially hosts microclimatic zones capable of
supporting a variety of habitats in relative stability (Qian and Ricklefs, 2000).
4.3 Demographic historyPopulations of E. argus and E. brenchleyi showed genetic imprints of demographic expansion as inferred by a negative Fs test, which was also supported by the multimodal mismatch distributions analysis, Rogers test of sudden expansion model and AMOVA (Rogers et al., 1996; Castoe et al., 2007; Nuñez et al., 2011). According to the inference of population expansion, E. argus has undergone historical slow growth or is stable in size and E. brenchleyi has undergone historical slow decline in size.
All populations of E. argus remained relatively stable or grew slowly during cold (positive δ18O values) and warm (negative δ18O values) periods, probably because the species survived in multiple refugia during cold periods (stadials). Occasional migration among expanded populations might have resumed during warmer periods (interstadials), and this intermittent gene flow between refugia might have made the species population size insensitive to population reduction resulting from cool climates (Li et al., 2009).
All populations of E. brenchleyi remained relatively stable or descended slowly through time during cold and warm periods, probably because there had been lower intermittent gene fl ow among populations in E. brenchleyi than in E. argus. Moreover, the expansion events beginning at 0.004 39 Ma for E. argus and 0.030 6 Ma for E. brenchleyi suggest that climate changes during the LGM may have differentially affected the demographic histories of these two species.
AcknowledgementsThis work was carried out in compliance with Chinese laws on animal welfare and research and was supported by grants from Chinese Ministry of Education (20070319006) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We thank Qilei HAO, Lihua LI, Longhui LIN, Hongxia LIU, Yaqing LIU, Laigao LUO, Guoqiao WANG, Xuefeng XU, Jianlong ZHANG, Qunli ZHANG and Wenge ZHAO for assistance during this research.
Arnold E. N., Arribas O., Carranza S.2007. Systematics of the Palaearctic and Oriental lizard tribe lacertini (Squamata:Lacerti dae:Lacertinae), with descriptions of eight new genera. Zootaxa, 1430: 1−86
Avise J. C.2000. Phylogeography: The history and formation of species. Cambridge: Harvard University Press
Bai W. N., Liao W. J., Zhang D. Y.2010. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytol, 188: 892−901
Bandelt H. J., Forster P., Röhl A.1999. Median-joining networks for inferring intraspecifi c phylogenies. Mol Biol Evol, 16: 37−48
Busack S. D., Lawson R., Arjo W. M.2005. Mitochondrial DNA, allozymes, morphology and historical biogeography in the Podarcis vaucheri (Lacertidae) species complex. Amphibia-Reptilia, 26: 239−256
Camargo A., Sinervo B., Sites J. W.2010. Lizards as model organisms for linking phylogeographic and speciation studies. Mol Ecol, 19: 3243−3488
Castoe T. A., Spencer C. L., Parkinson C. L.2007. Phylogeographic structure and historical demography of the western diamondback rattlesnake (Crotalus atrox): A perspective on North American desert biogeography. Mol Phylogenet Evol, 42: 193−212
Chen B. H.1991. Lacertidae. In Chen B. H. (Ed.), The Amphibian and Reptilian Fauna of Anhui. Hefei: Anhui Science and Technology Publishing House, 219−230
Clark A. M., Bowen B. W., Branch L. C.1999. Effects of natural habitat fragmentation on an endemic scrub lizard (Sceloporus woodi): An historical perspective based on a mitochondrial DNA gene genealogy. Mol Ecol, 8: 1093−1104
Ding G. H., Fu T. B., Ji X.2012. Caudal autotomy does not increase locomotor costs in the oriental leaf-toed gecko Hemidactylus bowringii. Asian Herpetol Res, 3: 141−146
Drummond A. J., Rambaut A.2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol, 7: 214
Dupanloup I., Schneider S., Excoffier L.2002. A simulated annealing approach to defi ne the genetic structure of populations. Mol Ecol, 11: 2571−2581
Elderkin C. L., Christian A. D., Vaughn C. C., Metcalfe-Smith J. L., Berg D. J.2007. Population genetics of the freshwater mussel, Amblema plicata (Say 1817) (Bivalvia: Unionidae): Evidence of high dispersal and post-glacial colonization. Conserv Genet, 8: 355−372
Excoffi er L., Laval G., Schneider S.2005. Arlequin Version 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online, 1: 47−50
Excoffier L., Smouse P. E., Quattro J. M.1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics, 131: 479−491
Forcina G., Panayides P., Guerrini M., Nardi F., Gupta B. K., Mori E., Al-Sheikhly O. F., Mansoori J., Khaliq I., Rank D. N., Parasharya B. M., Khan A. A., Hadjigerou P., Barbanera F.2012. Molecular evolution of the Asian francolins (Francolinus, Galliformes): A modern reappraisal of a classic study in speciation. Mol Phylogenet Evol, 65: 523–534
Fu J. Z.1998.Toward the phylogeny of the family Lacertidae: implications from mitochondrial DNA 12S and 16S gene sequences (Reptilia: Squamata). Mol Phylogenet Evol, 9: 118−130
Fu Y. X.1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics, 147: 915−925
Hall T. A.1999. BIOEDIT: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser, 41: 95−98
Hewitt G. M.1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc, 58: 247−293
Hewitt G. M.1999. Post-glacial re-colonization of European biota. Biol J Linn Soc, 68: 87−112
Hewitt G. M.2000. The genetic legacy of the quaternary ice ages. Nature, 405: 907−913
Hewitt G. M.2004. Genetic consequences of climatic oscillations in the Quatemary. Philos Trans R Soc B, 359: 183−195
Hipsley C. A., Himmelmann L., Metzler D., Müller J.2009. Integration of Bayesian molecular clock methods and fossilbased soft bounds reveals early Cenozoic origin of African lacertid lizards. BMC Evol Biol, 9: 151
Imbrie J., Hays J. D., Martinson D. G., McIntyre A., Mix A. C., Morley J. J.1984. The orbital theory of Pleistocene climate: support from a revised chronology of the marine δ18O record. In Berger A. L., Imbrie Hays J. H., Kukla G., Saltzman B. (Eds.), Milankovitch and Climate, Part I. Reidel: Dordrecht, 269−305
Jin Y. T., Brown R. P., Liu N. F.2008. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan Plateau. Mol Ecol, 17: 1971−1982
Ju L. X., Wang H. J., Jiang D. B.2007. Simulation of the Last Glacial Maximum climate over East Asia with a regional climate model nested in a general circulation model. Paleogeogr Paleoclimatol Paleoecol, 248: 376−390
Kimura M.1981. Estimation of evolutionary distances between homologous nucleotide sequences. Proc Natl Acad Sci USA, 78: 454−458
Li J. J., Fang X. M., Ma H. Z., Zhu J. J., Pan B. T., Chen H. L.1996. Geomorphological evolution of Late Cenozoic river from upper Yellow River and the uplift of Qinghai-Tibetan plateau on climatic changes. Sci China D, 26: 316−322
Li S. H., Yeung C. K. L., Feinstein J., Han L. X., Le M. H., Wang C. X.2009. Sailing through the Late Pleistocene: Unusual historical demography of an East Asian endemic, the Chinese Hwamei (Leucodioptron canorum canorum), during the last glacial period. Mol Ecol, 18: 622−633
Li Y. X., Xue X. X., Liu H. J.2004. Fossil lizards of Qinling Mountains. Vertebr Palasiatic,42: 171−176
Lin L. H., Ji X., Diong C. H., Du Y., Lin C. X.2010. Phylogeography and population structure of the Reevese’s butterfl y lizard (Leiolepis reevesii) inferred from mitochondrial DNA sequences. Mol Phylogenet Evol, 56: 601−607
Lin Z. H., Ji X.2005. Partial tail loss has no severe effects on energy stores and locomotor performance in a lacertid lizard, Takydromus septentrionalis. J Comp Physiol B, 175: 567−573
Lin Z. H., Qu Y. F., Ji X.2006. Energetic and locomotor costs of tail loss in the Chinese skink, Eumeces chinensis. Comp Biochem Physiol A, 143: 508−513
Malhotra A., Dawson K., Guo P., Thorpe R. S.2011. Phylogenetic structure and species boundaries in the mountain pitviper Ovophis monticola (Serpentes: Viperidae: Crotalinae) in Asia. Mol Phylogenet Evol, 59: 444−457
Martrat B., Grimalt J. O., Lopez-Martinez C., Cacho I., Sierro F. J., Flores J. A.2004. Abrupt temperature changes in the Western Mediterranean over the past 250 000 years. Science, 306: 1762−1765
Mayer W., Pavlicev M.2007. The phylogeny of the family Lacertidae (Reptilia) based on nuclear DNA sequences: Convergent adaptations to arid habitats within the subfamily Eremiainae. Mol Phylogenet Evol, 44: 1155−1163
Mori E., Sforzi A., Di Febbraro M.2013. From the Apennines to the Alps: recent range expansion of the crested porcupine Hystrix cristata L., 1758 (Mammalia: Rodentia: Hystricidae) in Italy. Ital J Zool, 80: 469−480
Newton A. C., Allnot T. R., Gillies A. C. M., Lowe A. J., Ennos R. A.1999. Molecular phylogeography, intraspecifi c variation and conservation of tree species. Trends Ecol Evol, 14: 140−145
Nuñez J. J., Woood N. K., Rabanal F. E., Fontanella F. M., Sites J. W.2011. Amphibian phylogeography in the Antipodes: Refugia and postglacial colonization explain mitochondrial haplotype distribution in the Patagonian frog Eupsophus calcaratus (Cycloramphidae). Mol Phylogenet Evol, 58: 343−352
Nylander J. A. A.2004. MRMODELTEST version 2.1. Computer program distributed by the author. Uppsala University, Uppsala.
Palumbi S. R.1996. Nucleic acids II: The polymerase chain reaction. In Hillis D. M., Moritz C., Mable B. K. (Eds.), Moleclar Systematics, 2ndEd. Massachusetts: Sinauer Associates, 205−247
Parsons T. J., Muniec D. S., Sullivan K, Woodyatt N, Alliston-Greiner R.,Wilson M. R., Berry D. L., Holland K. A., Weedn V. W., Gill P., Holland M. M.1997. A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet, 15: 363−368
Petit R. J., Duminil J., Fineschi S., Hampe A., Salvini D., Vendramin G. G.2005. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol, 14: 689–701
Qian H., Ricklefs R. E.2000. Large-scale processes and the Asian bias in temperate plant species diversity. Nature, 407: 180−182
Rambaut A., Drummond A. J.2007. Tracer, version 1.4. http:// beast.bio.ed.ac.uk/Tracer.
Rogers A. R., Fraley A. E., Bamshad M. J., Watkins W. S., Jorde L. B.1996. Mitochondrial mismatch analysis is insensitive to the mutational process. Mol Biol Evol, 13: 895−902
Ronquist F., Huelsenbeck J. P.2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19: 1572−1574
Ruiz-González A., Madeira M. J., Randi E., Abramov A. V., Davoli F., Gómez-Moliner B. J.2013. Phylogeography of the forest-dwelling European pine marten (Martes martes): New insights into cryptic northern glacial refugia. Biol J Linn Soc, 109: 1−18
Slatkin M.1996. In defense of founder-fl ush theories of speciation. Am Nat, 147: 493−505
Stenson A. G., Thorpe R. S.2003. Phylogeny, paraphyly and ecological adaptation of the colour and pattern in the Anolis roquet complex on Martinique. Mol Ecol, 12: 117−132
Sun Y. Y., Yang J, Ji X.2009. Many-lined sun skinks (Mabuya multifasciata) do not compensate for the costs of tail loss by increasing feeding rate or digestive effi ciency. J Exp Zool A, 311: 125−133
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S.2013.
MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol, 30: 2725−2729
Tian B., Liu R. R., Wang L. Y., Qiu Q., Chen K. M., Liu J. Q.2009. Phylogeographic analyses suggest that a deciduous species (Ostryopsis davidiana Decne., Betulaceae) survived in northern China during the Last Glacial Maximum. J Biogeogr, 36: 2148−2155
Urquhart J., Wang Y. Z., Fu J. Z.2009. Historical vicariance and male-mediated gene flow in the toad-headed lizards Phrynocephalus przewalskii. Mol Ecol, 18: 3714−3729
Weaver A. J., Eby M, Fanning A. F., Wiebe E. C.1998. Simulated infl uence of carbon dioxide, orbital forcing and ice sheets on the climate of the Last Glacial Maximum. Nature, 394: 847−853
Wu C., Zhang X. Q., Ma Y. H.1999. The Taihang and Yan mountains rose mainly in Quaternary. China Earthq Sci, 17: 1−7
Zamudio K. R., Savage W. K.2003. Historical isolation, range expansion, and secondary contact of two highly divergent mitochondrial lineages in spotted salamanders (Ambystoma maculatum). Evolution, 57: 1631−1652
Zhang Z. Y., Yu Q. W., Zhang K. X., Gu Y. S., Xiang S. Y.2003. Geomorphological evolution of Quaternary river from upper Yellow River and geomorphological evolution investigation for 1:250000 scale geological mapping in Qinghai-Tibet plateau. J China Univ Geosci, 28: 621−633
Zhao K. T.1999. Lacertidae. In Zhao E. M., Zhao K. T., Zhou K. Y. (Eds.), Fauna Sinica, Reptilia (Squamata: Lacertilia), Vol. 2. Beijing: Science Press, 219−242
Zhao Q., Liu H. X., Luo L. G., Ji X.2011. Comparative population genetics and phylogeography of two lacertid lizards (Eremias argus and E. brenchleyi) from China. Mol Phylogenet Evol, 58: 478−491
Zhao Q., Wang Z., Liu L. L., Zhao W. G., Ji X.2008. Selected body temperature, surface activity and food intake in tailed versus tailless Mongolian racerunners Eremias argus from three populations. Acta Zool Sinica, 54: 60−66
Zink R. M., Groth J. G., Vázquez-Miranda H., Barrowclough G. F.2013. Phylogeography of the California Gnatcatcher (Polioptila californica) using multilocus DNA sequences and ecological niche modeling: implications for conservation. Auk, 130: 449–458
*Corresponding author: Prof. Xiang JI, from College of Life Sciences, Nanjing Normal University, Jiangsu, China, with his research focusing on physiological and evolutionary ecology of reptiles.
E-mail: xji@mail.hz.zj.cn
Received: 25 October 2014 Accepted: 9 December 2014
 Asian Herpetological Research2014年4期
Asian Herpetological Research2014年4期
- Asian Herpetological Research的其它文章
- A New Species of the Genus Thermophis (Serpentes: Colubridae) from Shangri-La, Northern Yunnan, China, with a Proposal for an Eclectic Rule for Species Delimitation
- Group-spawning and Simultanous Polyandry of a Stream-dwelling Frog Feirana kangxianensis
- Can an Invasive Prey Species Induce Morphological and Behavioral Changes in an Endemic Predator? Evidence from a South Korean Snake (Oocatochus rufodorsatus)
- Food Habits and Distribution of the Lake Taal Sea Snake (Hydrophis semperi Garman 1881) and the Sympatric Little File Snake (Acrochordus granulatus Schneider 1799) in Lake Taal, Philippines
- Body Size and Reproductive Tactics in Varanid lizards
- Sexual Dimorphism in Mass of the Hindlimb Muscles of the Piebald Odorous Frog (Odorrana schmackeri)