
活血壮筋胶囊质量标准研究
2014-03-24 07:20杨玉霞刘竞研于秀华孙丹丹陈凡波尹建元赵芸浩孟勤
特产研究 2014年2期
杨玉霞,刘竞研,于秀华,孙丹丹,陈凡波,尹建元,赵芸浩,孟勤
(1.吉林大学药学院 a.天然药物化学教研室; b.药学实验中心,长春 130021; 2.东北师范大学附属中学,长春 130021)
活血壮筋胶囊由红花、血竭、桂枝、人参等11味中药组成,具有祛风活血、强腰壮筋的功效,临床用于筋骨疼痛、周身麻木、半身不遂、口歪眼斜[1]。活血壮筋胶囊是在不改变原来剂型制备工艺前提下,由活血壮筋丸改为胶囊剂[2]。为了进一步提高制剂质量控制水平,本试验增加了人参、桂枝、红花的薄层色谱鉴别方法,采用高效液相色谱法建立了血竭中血竭素高氯酸盐的含量测定方法。为活血壮筋胶囊的质量控制提供了依据,使制剂质量得到更好控制。
1 仪器与试药
1.1 仪器
LC-10ATvp型高效液相色谱仪(日本岛津公司),SPD-10Avp紫外检测器、CAMAG型薄层色谱扫描仪(瑞士CAMAG公司);薄层板(青岛海洋化工厂分厂); BT125D电子天平(0.01mg,德国赛得利斯有限公司);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
活血壮筋胶囊(批号120101-120303,西安澜泰药业有限公司);人参皂苷Rb1对照品(批号0703-200221)、人参皂苷Re对照品(批号110754-200218)、人参皂苷Rg1对照品(批号0703-200221)、人参对照药材(批号120917-200609)、桂皮醛对照品(批号110710-201217)、红花对照药材(批号120907-201609)、血竭素高氯酸盐对照品(批号0811-200203,中国北京药品生物制品检定院);乙腈为色谱纯;水为重蒸馏水;其他试剂为分析纯。
2 方法与结果
2.1 薄层鉴别
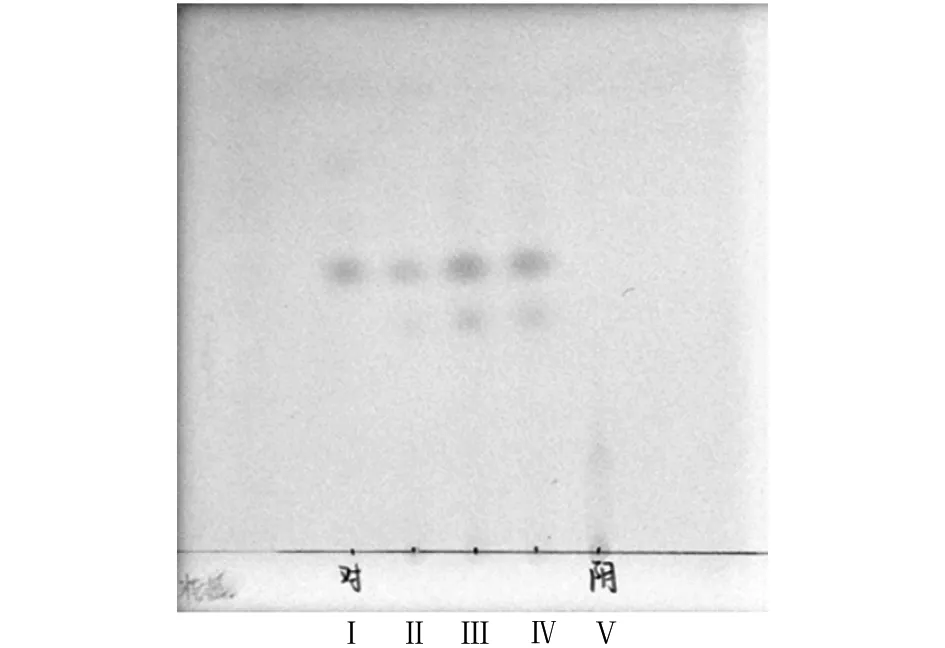
2.1.1人参的薄层鉴别取本品内容物4g,置具塞锥形瓶中,加甲醇30mL,超声处理30min,滤过,滤液蒸干,残渣加水20mL使溶解,用乙醚振摇提取3次,每次20mL,弃去,水层用水饱和的正丁醇振摇提取3次,每次20mL,合并提取液,用正丁醇饱和的氨试液洗涤2次,每次30mL,弃去,再用正丁醇饱和的水洗涤2次,每次20mL,正丁醇液蒸干,残渣加甲醇1mL使溶解,作为供试品溶液[3]。另取人参药材1g,同法制成对照药材溶液。再取人参皂苷Rb1、人参皂苷Re及人参皂苷Rg1对照品,加甲醇制成每1mL各含2mg的混合溶液作为对照品溶液。另按处方比例及工艺制法制备缺人参的阴性样品,同法制成阴性对照溶液。照薄层色谱法(中国药典2010年版一部附录ⅥB)试验,吸取对照药材溶液与对照品溶液各4μL,供试品溶液与阴性对照溶液各5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光及紫外光灯(365nm)下检视[4]。供试品色谱中,在与对照品色谱相应的位置上,日光下显相同的3个紫红色斑点,紫外光灯(365nm)下,显相同1个黄色和2个橙色荧光斑点。阴性样品在此对应位置上无斑点,即阴性无干扰,见图1。

2.1.2桂枝的薄层鉴别取本品内容物4g,置500mL圆底烧瓶中,加水200mL,按《中国药典》2010年版一部附录挥发油测定法,连接挥发油提取器,自提取器上端加水使充满刻度部分,再加入乙酸乙酯2mL,连接回流冷凝管,加热回流2h,分取乙酸乙酯层,用少量乙酸乙酯冲洗刻度部分,洗液并入乙酸乙酯液中,蒸干,残渣加无水乙醇1mL使溶解,作为供试品溶液。另取桂皮醛对照品,加乙醇制成每1mL含1μL的溶液,作为对照品溶液。另按处方比例及工艺制法制备缺桂枝的阴性样品,同法制成阴性对照溶液。照薄层色谱法(中国药典2010年版一部附录Ⅵ B)试验,吸取对照品溶液2μL,供试品溶液和阴性对照溶液各4μL,分别点于同一硅胶G薄层板上,以石油醚(30℃~60℃)-乙酸乙酯(17∶3)为展开剂,展开,取出,晾干,喷以2,4-二硝基苯肼乙醇试液[5]。供试品色谱中,在与对照品相应的位置上显相同颜色的斑点。阴性样品在此对应位置上无斑点,即阴性无干扰,见图2。
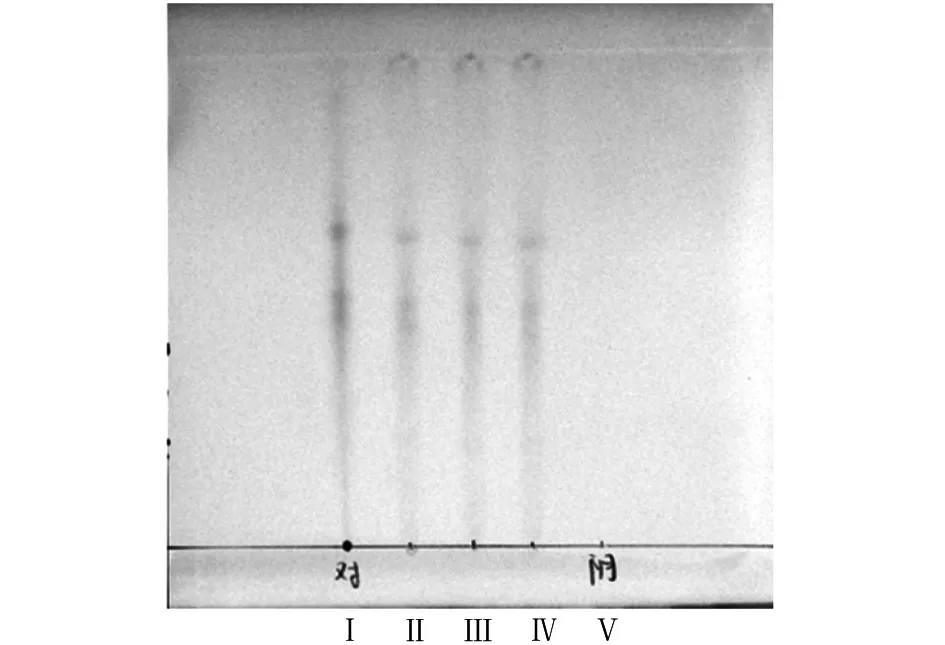
2.1.3红花的薄层鉴别取本品内容物4g,置锥形瓶中,加80%丙酮20mL,振摇15min,滤过,滤液蒸干,残渣加水15mL使溶解,用乙醚提取3次,每次20mL,弃去,水层挥尽乙醚后上已处理好的D101大孔树脂柱(内径1.5cm,柱高10cm),用60mL水洗脱,弃去,再用30%乙醇1000mL洗脱,收集洗脱液,蒸干,残渣加70%乙醇1mL使溶解,作为供试品溶液。另取红花对照药材,同法制成对照药材溶液。另按处方比例及工艺制法制备缺红花的阴性样品,同法制成阴性对照溶液。照薄层色谱法(中国药典2010年版一部附录ⅥB)试验,吸取供试品溶液与阴性对照溶液各5μL,对照品溶液2μL,分别点于同一硅胶H薄层板上,以乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶4)为展开剂,展开,取出,晾干[6]。供试品色谱中,在与对照药材色谱相应的位置上,显相同的斑点。阴性样品在此对应位置上无斑点,即阴性无干扰,见图3。
2.2 血竭含量测定
2.2.1色谱条件与系统适用性试验色谱柱:Shim-Pack VP-ODS C18柱(250mm×4.6mm,5μm),流动相:乙腈-0.05mol/L磷酸二氢钠溶液(43∶57),流速1.0mL/min,柱温40℃,检测波长440nm。理论板数按血竭素高氯酸盐峰计算应不低于3500。
2.2.2对照品溶液的制备精密称取血竭素高氯酸盐10mg,置25mL棕色量瓶中,加3%磷酸甲醇溶液使溶解并稀释至刻度,摇匀,精密量取1mL,置10mL棕色量瓶中,加甲醇至刻度,摇匀,即得每1mL含血竭素29μg的对照品储备液(血竭素重量=血竭素高氯酸盐重量/1.377)。
2.2.3供试品溶液的制备取本品内容物适量,研细,取约0.8g精密称定,置25mL棕色量瓶中,加入3%磷酸甲醇溶液20mL,超声处理30min,放至室温,加3%磷酸甲醇溶液至刻度,摇匀,滤过,精密吸取续滤液5mL,置10mL棕色量瓶中,加3%磷酸甲醇至刻度,摇匀,即得[7]。
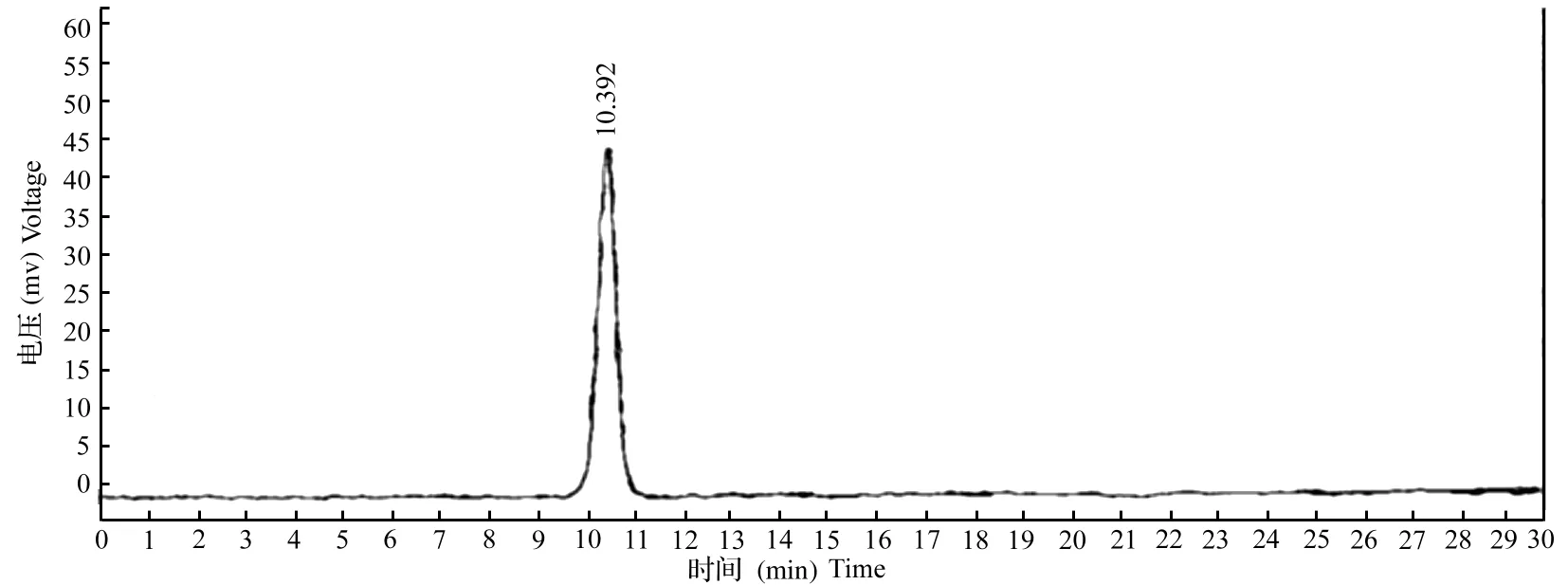
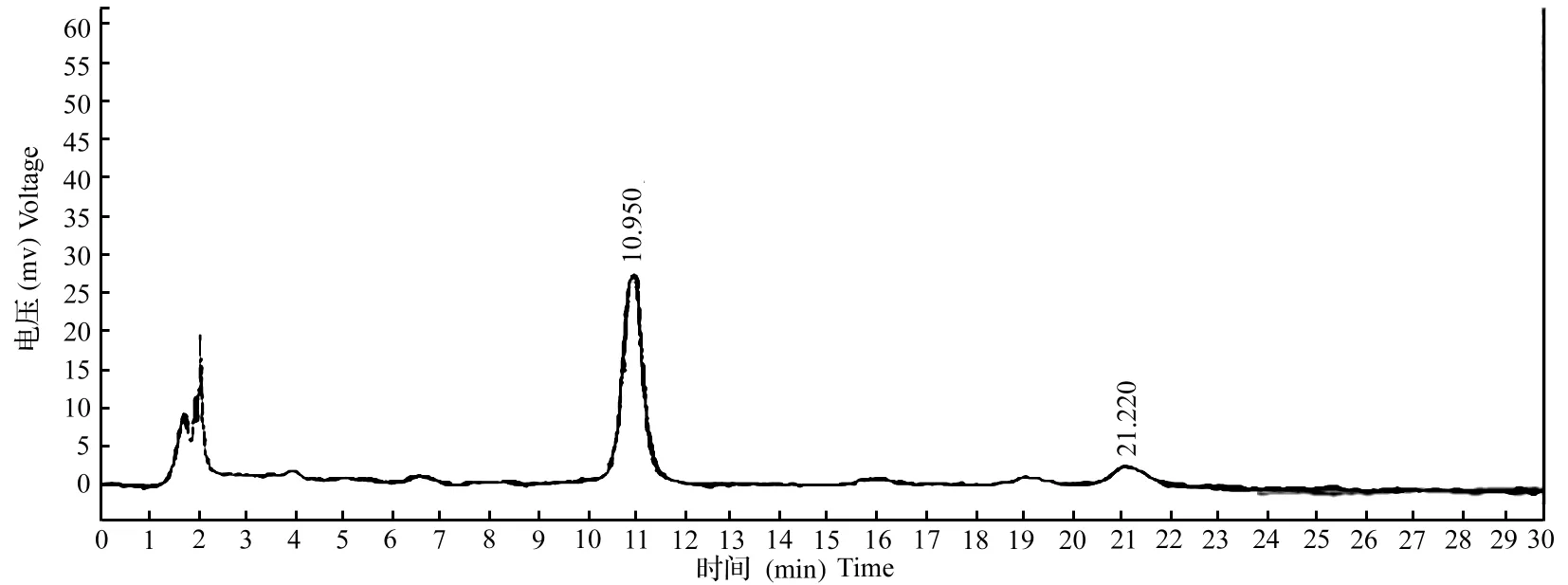
2.2.4空白试验按供试品溶液制备时取样量,折算成处方中除血竭外其他药材的取样量,按活血壮筋胶囊制法制成样品,再按供试品溶液制备方法制成空白溶液。分别取对照品溶液、供试品溶液、空白对照溶液各10μL,按“2.2.1”项下色谱条件测定,记录色谱图。对照品、供试品在相同的保留时间内有吸收峰,而空白对照溶液在此相应保留时间无吸收峰,说明阴性无干扰(图4、图5、图6)。
2.2.5线性关系的考察分别精密吸取对照品溶液4.0μL、8.0μL、10.0μL、12.0μL、16.0μL、20.0μL,注入液相色谱仪,测定。回归方程为Y=1 612 285.836X-4855.209 5,r=0.999 9。线性范围:0.080 64μg~0.403 2μg。
2.2.6稳定性试验依上述色谱条件,分别在0h、4h、8h、12h、16h、20h、24h测定同一批(批号:120105)供试品溶液,吸收峰峰面积RSD为0.67%。表明供试品溶液中血竭素高氯酸盐在24h内是稳定的。
2.2.7中间精密度试验按“2.2.2”项下方法制备的对照品溶液重复测定6次,吸收峰峰面积基本不变,RSD为0.28%(n=6),表明中间精密度良好。
2.2.8重现性试验取同一批号(批号:120105)样品,按“2.2.3”项下方法平行制备6份供试品溶液,依法进行测定,RSD为1.08%。
2.2.9加样回收率试验精密取已知含量的同一批号样品(批号:120105)6份,研细,取约0.5g(含量为0.525 4mg/g),精密称定,精密加入对照品溶液2mL(浓度为0.126mg/mL),按“2.2.3”项下方法制备供试品溶液,测定,计算回收率。结果见表1。试验结果表明,回收率平均值为97.89%,RSD值为1.40%,证明此方法可行。
表1回收率试验结果
Table1Thesamplerecoveryoftheprocedure
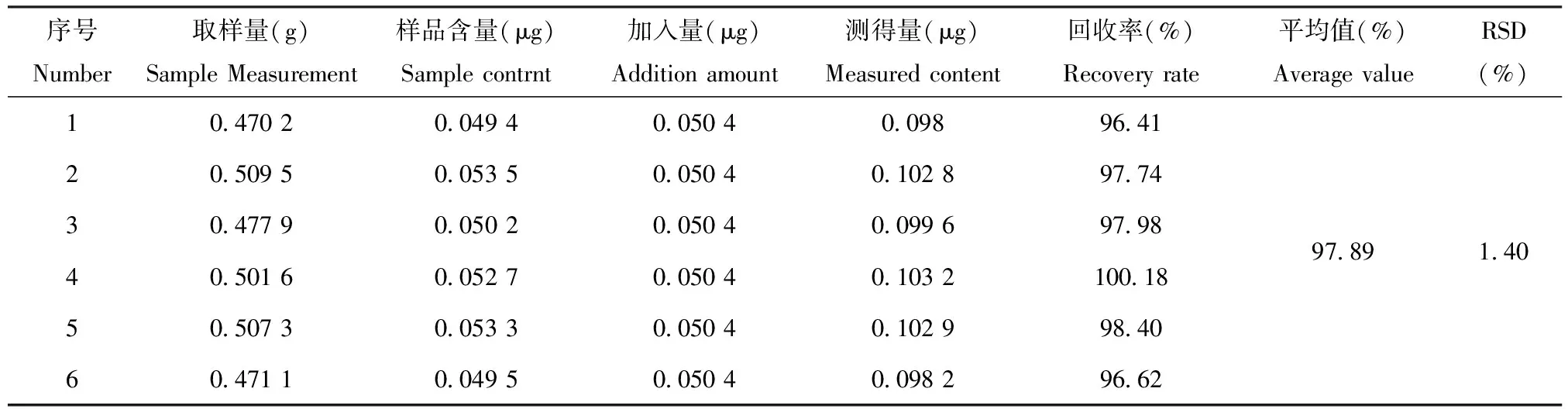
序号Number取样量(g)SampleMeasurement样品含量(μg)Samplecontrnt加入量(μg)Additionamount测得量(μg)Measuredcontent回收率(%)Recoveryrate平均值(%)AveragevalueRSD(%)1047020049400504009896412050950053500504010289774304779005020050400996979840501600527005040103210018505073005330050401029984060471100495005040098296629789140
2.2.10样品含量测定按“2.2.3”项下方法制备供试品溶液,取供试品溶液和对照品溶液各10μL,分别进样,测定。见表2。
表2样品含量测定结果
Table2ThecontentsofdracohodinperochlorateinHuoxuezhuangjincapsules
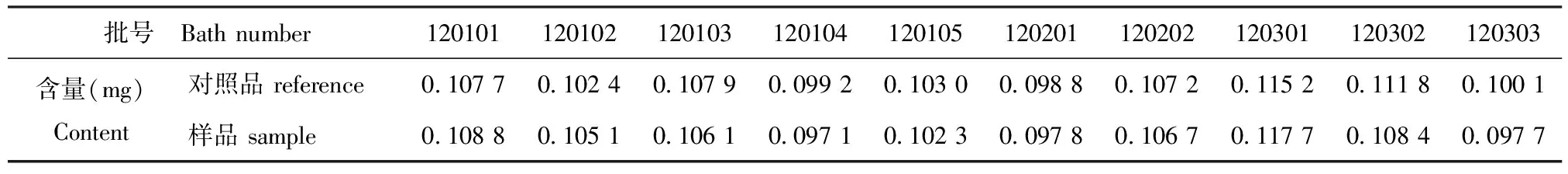
批号 Bathnumber120101120102120103120104120105120201120202120301120302120303含量(mg)Content对照品reference01077010240107900992010300098801072011520111801001样品sample01088010510106100971010230097801067011770108400977
根据10批样品测定的结果,另外考虑到药材产地、储藏及生产的波动。另因血竭中血竭素不十分稳定,怕光、怕热,所以按每粒平均含量0.105 3mg的80%计算,规定每粒含血竭以血竭素计不得少于0.085mg。
3 讨论
本研究采用薄层色谱法对处方中人参、桂枝和红花3味药进行了定性鉴别,因其处方成分复杂,为了排除干扰,在人参及红花的检测项中,将前处理步骤进行了优化,而桂枝的主要成分桂皮醛为挥发性成分[7],采用挥发油提取法,方法简便,专属性强。结果薄层斑点清晰,重现性好,阴性无干扰,可以列入活血壮筋胶囊的质控标准。
血竭素为色原酮类化合物,见光易分解,性质不稳定,因此本试验的对照品和样品为避免强光照射均储存于棕色瓶中。以3%磷酸甲醇溶液作为溶剂,可使样品中的血竭素转变为血竭素磷酸盐,增强稳定性[8]。本试验采用高效液相色谱法建立了血竭素含量测定方法,该方法经方法学验证,重复性及回收率良好,可以列入活血壮筋胶囊的质控标准,亦可为中药复方中血竭素测定提供参考。
[1]李彦超,王艳宋,汉敏,等.RP-HPLC法测定活血壮筋丸(丹)中血竭素的含量[J].南京中医药大学学报,2011,7(27):396-397.
[2]刘绪林.活血壮筋胶囊的质量标准研究[J].中国医药指南,2012,12(10):70-72.
[3]杨瑞瑞,李雪,乔蓉霞,等.展筋活血散质量标准研究[J].中国药品标准,2013,14(4):251-255.
[4]汪冰,林永强,徐丽华.芪参胶囊的质量标准研究[J].中国药事,2013,27(6):592-596.
[5]ZGB2011-1,中国药品标准[S].
[6]国家药典委员会.中华人民共和国药典[K].北京:中国医药科技出版社,2010:141.
[7]许源,宿树兰,王团结,等.桂枝的化学成分与药理活性研究进展[J].中药材,2013,36(4):674-677.
[8]窦金凤,曹云飞.HPLC法测定活血壮筋丸中血竭素的含量[J].现代仪器,2009,(2):32-33.
猜你喜欢
昆明医科大学学报(2021年1期)2021-02-07
海峡姐妹(2019年8期)2019-09-03
中成药(2018年5期)2018-06-06
小天使·一年级语数英综合(2017年11期)2017-12-05
海峡科技与产业(2016年3期)2016-05-17
华人时刊(2016年13期)2016-04-05
中国生化药物杂志(2015年4期)2015-07-07
中国当代医药(2015年8期)2015-03-01
中国药业(2014年24期)2014-05-26
中国合理用药探索(2014年11期)2014-03-11
