
水合三氯乙醛前体物的分子量分布和荧光特性
2014-03-20 02:19蔡广强刘丽君张金松卢小艳
净水技术 2014年5期
蔡广强,刘丽君,张金松,,卢小艳,徐 荣
(1. 哈尔滨工业大学深圳研究生院,广东深圳 518055;2. 深圳市水务〈集团〉有限公司,广东深圳 518031)
水合三氯乙醛(chloral hydrate,CH)在氯化消毒副产物中含量仅次于三卤甲烷(trihalomethanes,THMs)和卤乙酸(haloacetic acids,HAAs),为第三大类消毒副产物(disinfection by-products,DBPs),但CH 对人体的危害性远超过THMs 和HAAs[1-4]。在加拿大、美国、澳大利亚和我国北京等[5-8]出厂水中均有检出CH,其最大浓度均超过我国《生活饮用水卫生标准》(GB 5749—2006)[9]和世界卫生组织饮用水水质准则[10]的标准限值(10 μg/L),具有较高的超标风险。
水源中溶解性有机物(dissolved organic matter,DOM)被认为是CH 的主要前体物来源[3],为探寻CH 主要前体物种类,以南方某市一主要水源水库为研究对象,采用不会破坏DOM 结构的超滤技术[11]对夏季代表性水样中的DOM 进行分子量(Molecular weight,MW)分级,分析不同分子量区间有机物的CH 生成势(chloral hydrate formation potential,CHFP)和比CH 生成势(special chloral hydrate formation potential,SCHFP),结合三维激发-发射矩阵(three-dimensional excitation and emission matrix,3D-EEM)荧光光谱技术对不同分子量区间的DOM 组分进行表征,找出CH 主要前体物的分子量分布区间。与20 种常见的氨基酸、牛血清白蛋白(bull serum albumin,BSA)、鲱鱼精DNA、糖类、脂肪酸、富里酸、腐殖酸等模式化合物的氯化结果对比分析,最终确定CH 的主要前体物种类与性质,为水处理中有效控制CH 的生成提供技术指导,从而保障饮用水的安全生产。
1 材料与方法
1.1 溶液的配制
试验过程中所用试剂均采用优级纯化学试剂,所用水来自NANO pure 超纯水系统(Thermo Scientific 7146)。氯的储备液(1 000 mg/L,以Cl2计)采用有效氯大于5%的次氯酸钠(NaOCl)溶液配 制 而 成,以 DPD/FAS 滴 定 法 进 行 标 定[12]。0.01 mol/L磷酸盐缓冲液(pH 为7)采用磷酸二氢钾和磷酸氢二钾配制而成。20 种氨基酸、BSA、鲱鱼精DNA、葡萄糖、淀粉、脂肪酸、富里酸、腐殖酸等配制溶液浓度为1 mg/L(以DOC 计)。
1.2 水样处理与保存
水样于2013 年3 月~12 月采自我国南方某水库水源。每月中旬进行采样,采样频率为每月1 次,采用有机玻璃采水器于该水源取水口5 米深处进行取样,水样置于事先清洗干净的聚四氟乙烯塑料桶中,水样取回后立即采用0.45 μm 玻璃纤维滤膜(GF/C,Whatman,UK)过滤,若不能即刻进行试验,水样应放置在4 ℃冰箱中避光保存,水样保存不应超过7 d。
1.3 超滤分离试验
采用超滤杯(Amicon,Millipore 8400)与YM 10,YM 3 和YM 1 的超滤膜(Amicon,Millipore)(孔径分别为10、3、1 kDa)组成的超滤装置进行水样分离,试验流程如图1 所示。
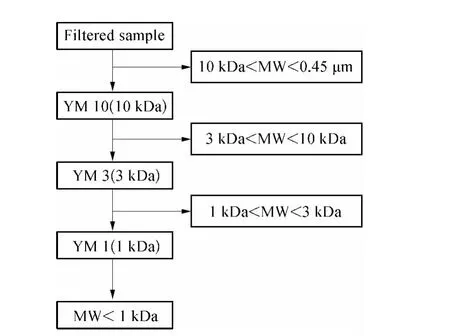
图1 超滤分离试验流程Fig.1 Schematic Diagram of Ultrafiltration Separation
分离过程中,采用高纯氮气加压,压力为0.1 ~0.5 MPa,压力大小与膜孔径成反比。首先将1 000 mL滤后水先通过YM 10 超滤膜,当样品体积约166 mL(水样初始体积的1/6)时停止过滤,同时收集膜出水用于后续试验;之后将200 mL(水样初始体积的1/5)超纯水陆续加入超滤杯继续加压超滤试验直到杯中样品体积恢复到166 mL 左右,以去除分子量小于膜截留分子量的有机物,同时将膜出水舍弃。最后,将杯中保留水样体积用超纯水稀释至原始样品体积(1 000 mL),以复原初始水样中该分子量区间有机物的DOC 水平,从而得到10 kDa <MW <0.45 μm水样。同样的分离方法用于YM 3 和YM 1超滤膜,最终得到四部分不同分子量区间有机物,即MW <1 kDa、1 kDa <MW <3 kDa、3 kDa <MW <10 kDa 和10 kDa <MW <0.45 μm[13]。
1.4 DOM 的3D-EEM 荧光光谱表征
为避免pH、温度、DOC、溶剂的极性等对检测结果的影响,荧光测定前应将所有样品调为统一试验条件[14,15]。所有样品DOC 均调为5 ±0.2 mg/L,并用HCl 调节至pH =3 以防止测量过程中沉淀的产生。用1 mol/L 的KCl 调节样品的KCl 浓度为0.01 mol/L,并以0.01 mol/L KCl 作为空白以校正试验过程中水样的拉曼散射。
3D-EEM 荧光光谱使用日本日立F-7000 荧光分光光度计进行测定,响应时间为自动,扫描速度为1 200 nm/min,其中激发波长(excitation wavelength,Ex)从200 nm 扫描到400 nm,间隔为5 nm,发射波长(emission wavelength,Em)从280 nm扫描到500 nm,间隔为2 nm,采用软件Origin 8.5 处理得到的3D-EEM 数据。由EEM 数据计算荧光指数(Fluorescence index,FI),FI 为450 nm 和500 nm发射波长在370 nm 激发波长下所对应的荧光强度的比值。FI 较大(~1.9)表明水体中以内源性有机物(来源于微生物)为主,FI 较小(~1.4)表明水体中以外源性有机物(来源于土壤)为主[16]。
荧光计算根据Chen 等研究结果[17],对EEM 进行分区,各区域用荧光体积积分法(FRI)进行定量计算。
区域“i”和总的荧光体积计算公式如下。
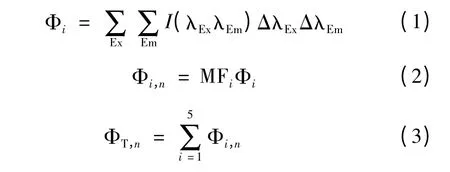
其中Φi——区域“i”的荧光体积;
Φi,n——标准化后的区域“i”荧光体积;
ΦT,n——5 个区域的荧光体积之和;
MFi——区域“i”的多重复性因子,即各个区域面积的倒数,以对Φi进行标准化,消除肩峰的影响,更有利于各个区域的比较;
ΔλEx——激发波长间隔,5 nm;
ΔλEm——发射波长间隔,2 nm;
I(λExλEm)——每一个激发-发射波长对所对应的荧光强度。
1.5 CH 生成势试验
参考USEPA 的消毒副产物生成势(disinfection by-products formation potential,DBPFP)测定标准方法[12],氯化前进行耗氯量预试验,将反应7 d 后余氯为3 ~5 mg/L 的加氯量作为初始加氯量。用HCl和NaOH 调节各水样pH 为7. 0 ± 0. 2,并加入0.01 mol/L的磷酸盐缓冲液维持pH 稳定,将水样置于250 mL 带有聚四氟乙烯螺旋瓶盖的琥珀色玻璃瓶中,250 mL/瓶,25 ±2 ℃条件下避光氯化培养7 d,最后采用亚硫酸钠(Na2SO3)作为终止剂进行脱氯。每个水样均在7 d 的氯化培养前后检测CH 浓度,两者之差即为CHFP。CHFP 与相应的DOC 之比得到SCHFP,表征单位DOC 的CH 生成能力。
1.6 试验仪器与分析方法
浊度采用HACH-2100Q 浊度仪检测;pH 采用Mettler Toledo-SG68 便携式pH 计检测;UV254采用VARIAN CARY50 型号紫外-可见分光光度计检测;TOC 采用Sievers 5310C 总有机碳测定仪;余氯采用HACH PC II 58700-00 便携式余氯仪检测;CH 测定参照GB/T 5750—2006[9]中的测定方法。
2 试验结果
2.1 不同月份原水的基本水质参数与CHFP
不同月份原水基本水质参数及CHFP 如表1所示。
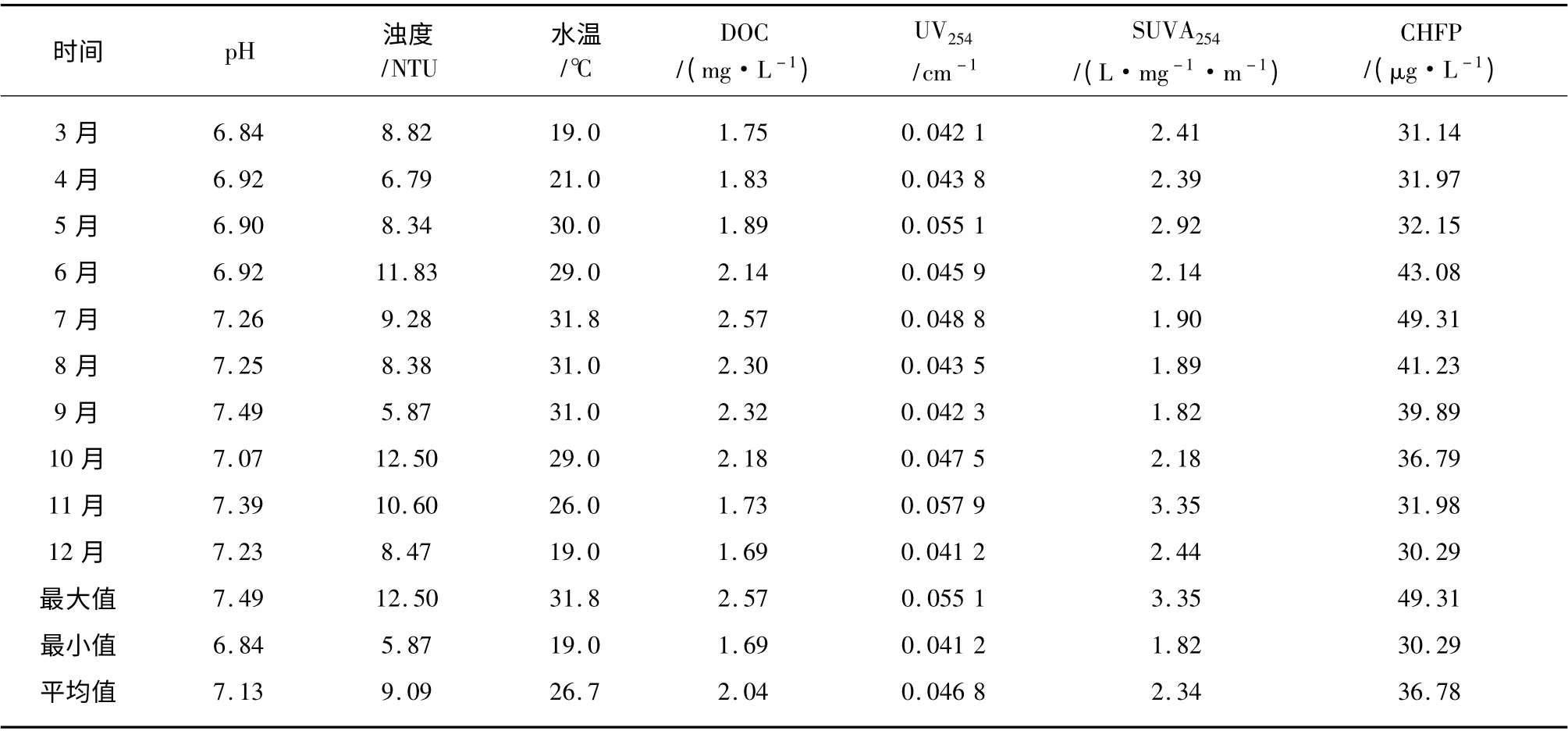
表1 不同月份原水基本水质参数及CHFPTab.1 Water Quality and CHFP of Raw Water Samples in Different Months
由表1 可知6 月~10 月即夏季和初秋时节CHFP 相对较高,这与水温和DOC 的变化趋势基本一致,说明CH 前体物含量与环境温度和DOM浓度具有一定的相关性。SUVA254反映水样中单位DOC 的芳香性程度,由于在6 月~10 月原水的SUVA254值相对较低,特别是在7 月~9 月,原水的SUVA254值均小于2 L/mg·m,说明此时原水中有机物主要以亲水性非腐殖类物质为主[18,19],由此推测CH 的主要前体物可能为亲水性的非腐殖类物质。
2.2 夏季水样中不同分子量区间DOM 组成及CH 生成
2.2.1 不同分子量区间DOM 组成
由于CH 前体物含量在夏季和初秋相对较高,故在夏季的代表性月份(2013 年7 月)对水样进行分子量分级试验,原水及分离后有机物各组分DOC组成如图2 所示。
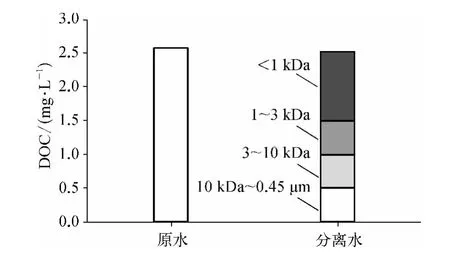
图2 原水及各分子量区间DOM 的DOCFig.2 DOC of Raw Water and DOM Fractions
由图2 可知,以DOC 来表示分离试验DOM 的回收率(分离后各分子量区间DOM 的DOC 之和与原水DOC 之比)为98. 05%,损失的DOM 部分(1.95%)可能是由于分离过程中膜对DOM 的吸附所致,相关研究结果[20]表明分离试验误差<10%均可接受,说明此次分离试验结果可靠。
分离后各分子量区间DOM 中,MW <1 kDa DOM 所占比例最大,为40. 87%;其余依次为1 kDa <MW <3 kDa DOM (19. 88%)、10 kDa <MW <0.45 μm DOM (19. 68%)、3 kDa <MW <10 kDaDOM (19.48%),夏季水样中以MW <1 kDa的小分子有机物为主,且MW >1 kDa 三部分DOM所占比例相差不大。
2.2.2 各分子量区间DOM 组分的CHFP 及SCHFP
各分子量区间DOM 组分的CHFP 和SCHFP 如图3 所示。
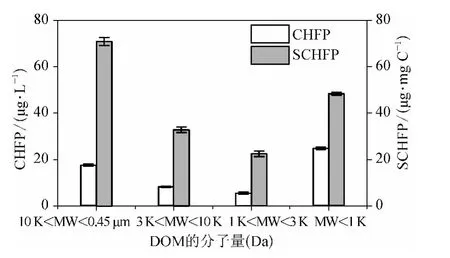
图3 各分子量区间DOM 组分的CHFP 和SCHFPFig.3 CHFP and SCHFP of DOM Fractions
由图3 可知各分子量区间DOM 组分的CHFP从大到小依次为MW <1 kDa (24. 81 μg/L)、10 kDa <MW <0. 45 μm (17. 49 μg/L)、3 kDa <MW <10 kDa (8.16 μg/L)和1 kDa <MW <3 kDa(5.50 μg/L),即MW <1 kDa DOM 为CH 的主要前体物,与MW <1 kDa DOM 所占DOC 比例最大一致;SCHFP 与CHFP 有所差别,从大到小依次为:10 kDa <MW <0. 45 μm (70. 65 μg/mg C)、MW <1 kDa (48. 17 μg/mg C)、3 kDa <MW <10 kDa (32.64 μg/mg C)和1 kDa <MW <3 kDa(22.47 μg/mg C);10 kDa <MW <0.45 μm DOM具有最大的CH 生成能力,说明10 kDa <MW <0.45 μm DOM 与氯的反应活性较高,更易生成CH,而CH 的主要前体物(MW <1 kDa DOM)并不具备最大的CH 生成能力,相比10 kDa <MW <0.45 μm 的DOM 较难生成CH,这可能与不同分子量区间有机物结构相关。
2.3 不同分子量区间DOM 的3D-EEM 荧光光谱表征
不同分子量区间DOM 组分的3D-EEM 荧光光谱如图4 所示。
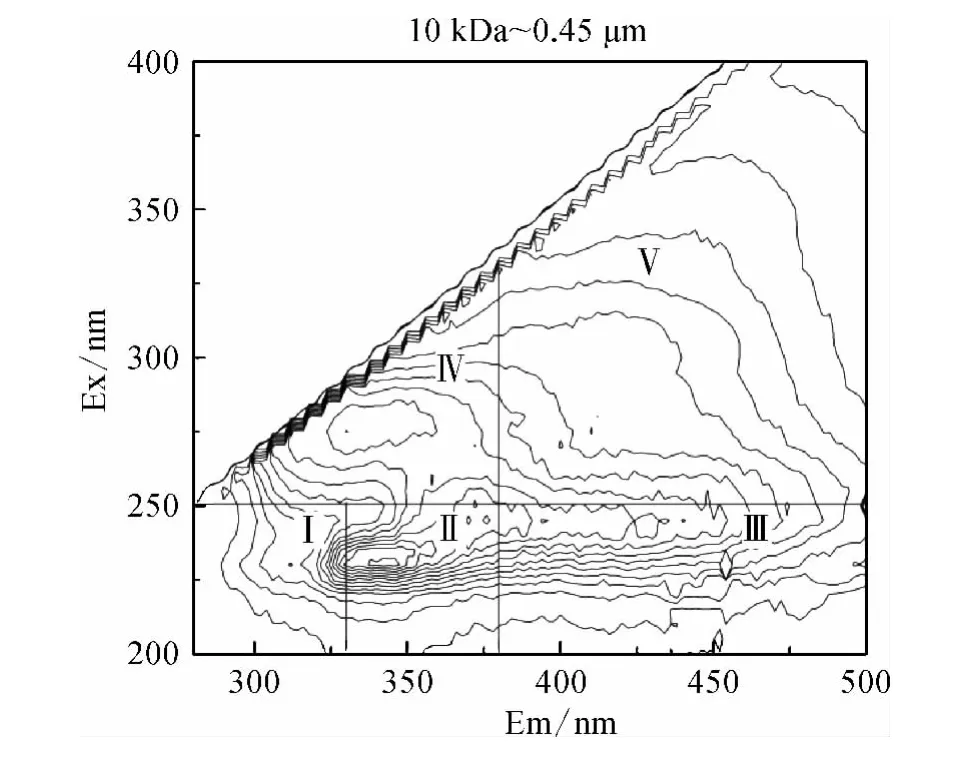
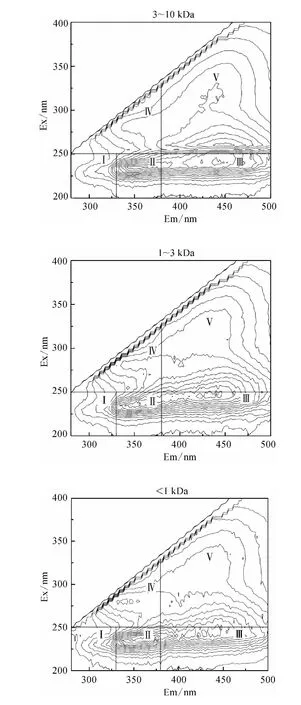
图4 不同分子量区间DOM 组分的3D-EEM 荧光光谱Fig.4 3D-EEM Fluorescence Spectra of DOM Fractions
Chen 等[17]的研究表明3D-EEM 荧光光谱根据模式化合物的荧光特性共划为五个区域,区域Ⅰ(λEm<330 nm,λEx<250 nm)和区域Ⅱ(330 nm <λEm<380 nm,λEx<250 nm)表征类芳香性蛋白质类物质;区域Ⅲ(λEm>380 nm,λEx<250 nm)表征类富里酸类物质;区域Ⅳ(λEm<380 nm,λEx>250 nm)表征类微生物代谢产物类物质;区域Ⅴ(λEm>380 nm,λEx>250 nm)表征类腐殖酸类物质。
由图4 可知所有分子量区间DOM 组分在区域Ⅰ均没有明显的特征峰出现,均以肩峰形式呈现,而各有机物在区域Ⅱ和区域Ⅲ均出现明显的特征峰;10 kDa <MW <0.45 μm DOM 在区域Ⅳ有明显的特征峰,其他三个分子量区间的DOM 在区域Ⅳ则以肩峰形式出现;只有3 kDa <MW <10 kDa DOM 在区域Ⅴ有特征峰出现,其他三个区间的有机物在区域Ⅴ则以肩峰形式出现。
结合2.2.2 中SCHFP 结果,所有分子量区间DOM 均能形成CH,且在区域Ⅱ和区域Ⅲ均出现特征峰,说明类芳香性蛋白质和类富里酸类物质可能是CH 的前体物,而10 kDa <MW <0.45 μm DOM的SCHFP 最大,且在区域Ⅳ具有单独的特征峰,3 kDa <MW <10 kDa DOM 的SCHFP 较小,且在区域Ⅴ单独有明显的特征峰,说明CH 的主要前体物可能为类微生物代谢产物类物质而非类腐殖酸类物质。
DOM 不同分子量组分ΦT,n及分布如表2 所示

表2 DOM 不同分子量组分ΦT,n及分布Tab.2 Distribution and ΦT,n of DOM Fractions by FRI Analysis
由表2 可知ΦT,n从大到小依次为MW <1 kDa、1 kDa <MW <3kDa、10 kDa <MW <0. 45μm、3 kDa <MW <10 kDa,与各分子量区间DOM 组分的SCHFP 没有一致趋势出现,表明CH 的生成主要取决于DOM 的荧光特性而不是DOM 的荧光总量。各分子量区间DOM 组分在区域Ⅰ、Ⅱ、Ⅲ三个区域的荧光体积分布与各DOM 组分的SCHFP 大小分布趋势基本一致,由此推测芳香性蛋白质和溶解性的微生物代谢产物可能是CH 的主要前体物;而DOM各分子量区间组分在区域Ⅲ和区域Ⅴ的荧光体积分布与各有机物组分的SCHFP 大小分布基本呈相反趋势,由此推测类富里酸类物质和类腐殖酸类物质都不是CH 的主要前体物,尽管各分子量区间DOM在区域Ⅲ均呈现特征峰。
由表2 可知DOM 各分子量区间组分的FI 均在1.4 ~1.9,说明各有机物组分均包含外源性物质和内源性物质,但3 kDa <MW <10 kDa DOM 的FI 最小(1.55),则此分子量区间有机物主要由外源性物质组成,如腐殖酸、富里酸等,而10 kDa <MW <0.45 μm DOM 的FI 值最大(1.82),表明此分子量区间有机物中蛋白质类等内源性物质比例相对较高。
2.4 模式化合物的CH 生成量
为了验证2.3 中对试验结果的推测,选取20 种常见的氨基酸、BSA(代表蛋白质)、鲱鱼精DNA(代表DNA)、葡萄糖、淀粉(代表多糖)、脂肪酸等生物来源的有机物以及富里酸、腐殖酸等有机物进行氯化试验,结果如图5 所示。
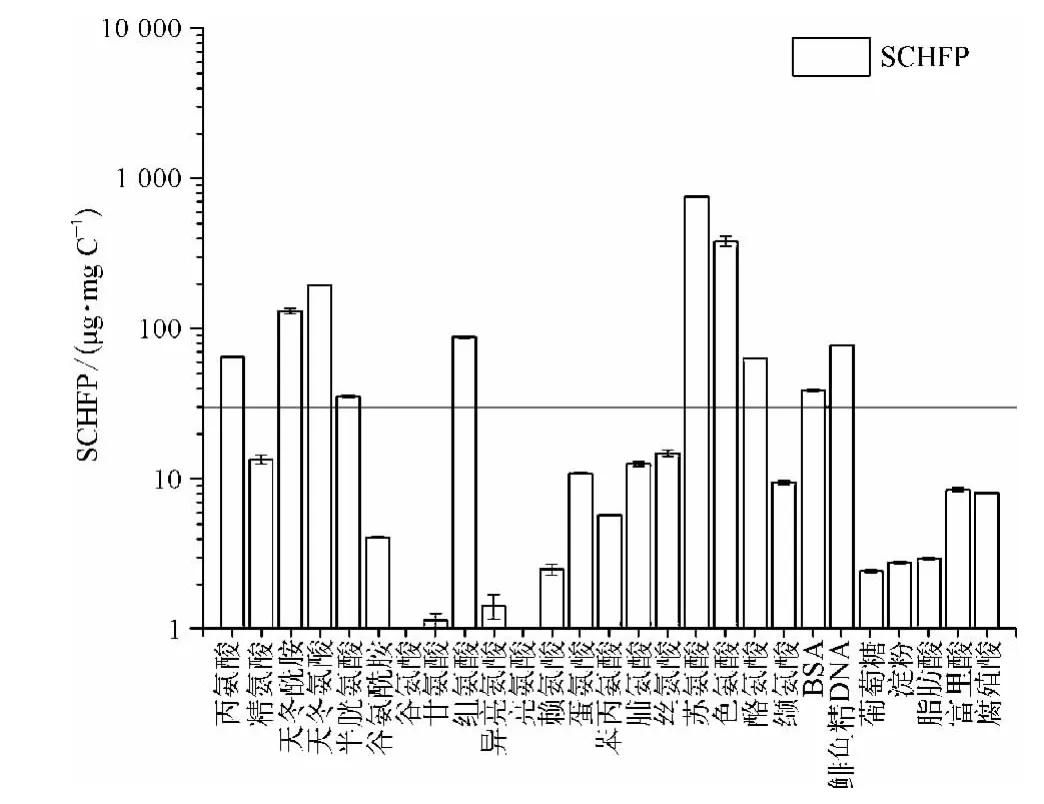
图5 模式化合物的CH 生成量Fig.5 CH Yields of Model Compounds
由图5 可知丙氨酸、天冬酰胺、天冬氨酸、半胱氨酸、组氨酸、苏氨酸、色氨酸、酪氨酸、BSA、DNA 等的CH 生成能力较强,均大于30 μg/mg C;其他12种氨基酸、以葡萄糖和淀粉为代表的糖类、脂肪酸、腐殖酸、富里酸等有机物的CH 生成能力较弱,不是CH 的主要前体物。其中富里酸和腐殖酸的SCHFP均为8 μg/mg C 左右,则丙氨酸、天冬酰胺、天冬氨酸、半胱氨酸、组氨酸、苏氨酸、色氨酸、酪氨酸、蛋白质、DNA 等的CH 生成能力分别是其生成能力的8、16、24、4、11、95、47、8、5 和10 倍。由此可知丙氨酸、天冬酰胺等氨基酸、蛋白质等含氮类有机物是CH 的主要前体物质。由于蛋白质主要由氨基酸组成,故研究氨基酸生成CH 的途径有利于CH 机理研究,相关研究[21,22]表明氨基酸生成CH 的大致反应途径如图6 所示。
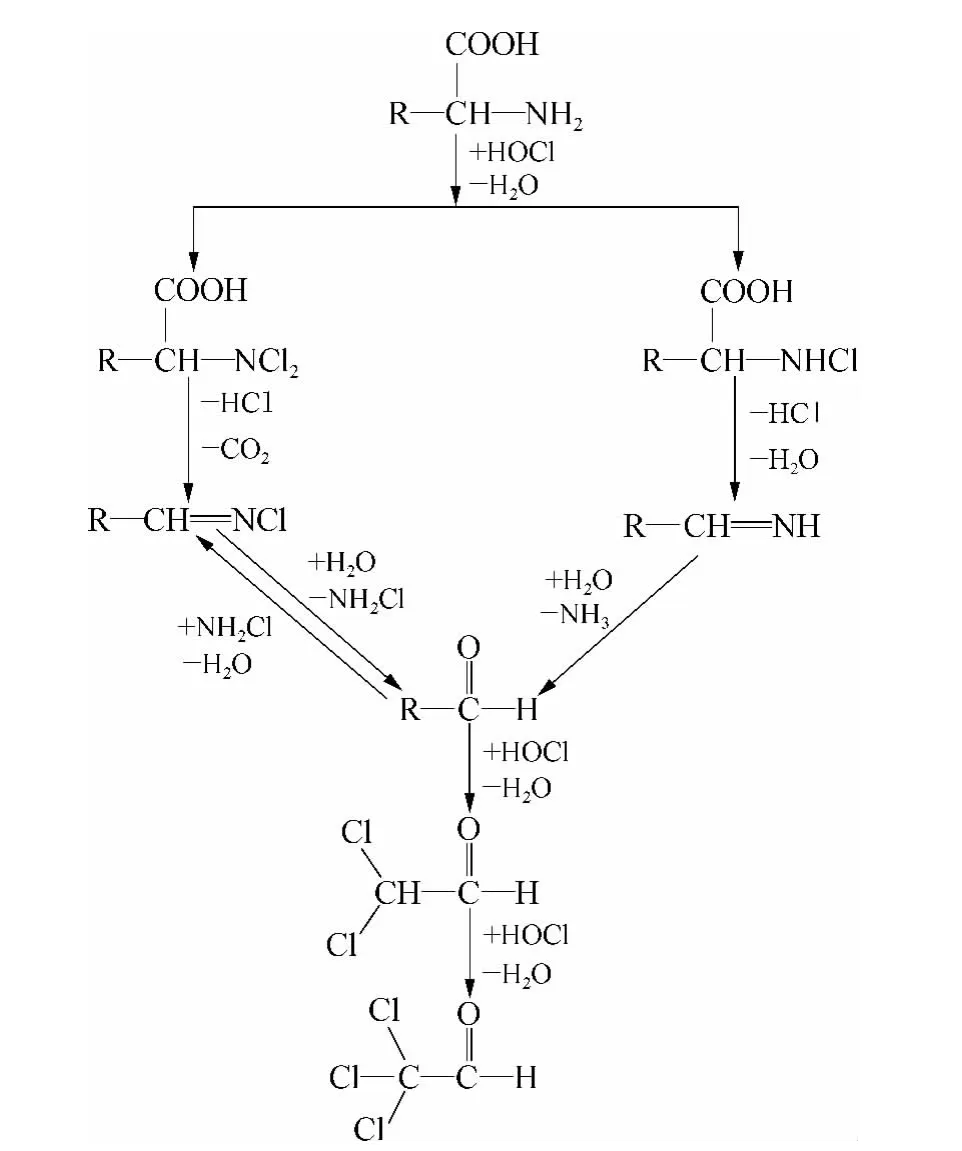
图6 氨基酸氯化过程中生成CH 的反应路径ig.6 CH Formation Pathway of AAs during Chlorination
氨基酸生成CH 的大致反应过程即经过取代、消去、水解等反应过程生成醛类的中间产物(RCHO),之后经过HOCl 氧化等反应过程生成二氯乙醛(dicholoroacetaldehyde,DCA),最终生成CH。
3 讨论
CH 的前体物含量呈现明显的季节性变化趋势,夏季、初秋的CH 前体物含量相对较高,这可能是由于高温使得藻类暴发频繁,大量藻类分泌物进入水体所致;此外,由于夏季雨量较大,雨水也可能带入一定量的有机物进入水体中,进而成为CH 的前体物。Henderson、Chang 等[23,24]对不同藻体有机质的表征结果表明,藻类有机质主要由SUVA254较低的亲水性物质组成,SUVA254一般为2 L/mg·m,这与我们6 月~10 月的原水SUVA254值较低基本一致。说明在夏季、初秋时,原水可能由于藻类暴发或者径流污染带入一定量的有机物,使得CH 前体物含量大量增加。
夏季代表性月份的水样中以MW <1 kDa 和10 kDa <MW <0. 45 μm DOM 为CH 的主要前体物,两者占CH 前体物含量的75. 55%;这两部分DOM 的SUVA254和3D-EEM 光谱表征结果都显示这两部分有机物的蛋白质、氨基酸等亲水性有机物含量较高。方晶云等[22]的研究表明藻源性有机物的分子量分布以MW <1 kDa 和10 kDa <MW <0.45μm这两部分有机物为主,而藻源性有机物主要由蛋白质、氨基酸等亲水性的DON 类物质组成[24],说明水样中MW <1 kDa 和10 kDa <MW <0.45 μm两部分DOM 可能来源于藻类的分泌物,这与相关的研究结论CH 前体物与DOM 类有机物相关一致[25]。各分子量区间有机物进行3D-EEM 荧光光谱表征的结果也表明CH 的主要前体物可能为芳香性蛋白质(区域Ⅱ)和微生物代谢产物(区域Ⅳ),而微生物代谢产物又多为芳香性蛋白质、氨基酸等有机物组成[17,26],同时还包括DNA、脂肪酸等生物源类物质。在Ex/Em 波长对为230/340 nm/nm(区域Ⅱ)和280/340 nm/nm(区域Ⅳ)的荧光峰表征有机氮丰富的有机物,说明CH 的前体物应该为含氮丰富的蛋白质、氨基酸等物质[17]。
丙氨酸、天冬酰胺、天冬氨酸、半胱氨酸、组氨酸、苏氨酸、色氨酸、酪氨酸、蛋白质、DNA 等有较大的CH 生成量,主要原因可能是丙氨酸的R 基为甲基,而甲基极易与HOCl 发生取代反应,产生CH;而天冬酰胺、天冬氨酸、半胱氨酸、组氨酸、苏氨酸、色氨酸、酪氨酸等7 种氨基酸的R 基是较强的电子供体,尤其是组氨酸、色氨酸和酪氨酸分别属于杂环和芳香族氨基酸,从而使得氨基更易于被HOCl 氧化生成醛基而生成CH;蛋白质由氨基酸组成,DNA 也属于杂环类有机物,可能易与氯反应生成CH。Hureiki 等[21]的研究也表明天冬酰胺、天冬氨酸、组氨酸、色氨酸和酪氨酸的总有机卤素生成势(total organic halogen formation potential,TOXFP)较大,均大于1 M/M AAs,而半胱氨酸和苏氨酸的TOXFP 也在0.5 M/M AAs 左右,与上述试验结果基本吻合。Trehy 等[27]对天冬氨酸、色氨酸、酪氨酸等三种氨基酸的氯化试验也表明这三种氨基酸均能产生大量的CH,成为主要的CH 前体物。而其他12 种氨基酸、糖类、富里酸和腐殖酸等并没有产生大量的CH,说明有机物的结构对于CH 生成具有较大影响[21]。
以上结果显示,夏季水样中CH 的前体物为MW <1 kDa 和10 kDa <MW <0.45 μm DOM 为主,且蛋白质、氨基酸等亲水性物质成为主要CH 前体物组分。由于强化混凝有利于去除大分子有机物[28],而生物处理则有利于去除小分子的亲水性有机物[29],因此,建议水厂处理过程中采用强化混凝、生物处理等来去除CH 的前体物,以控制CH 的生成。
4 结论
(1)夏、初秋时节,CH 的前体物含量较高,来源可能主要是藻类的分泌物和径流引入的污染物质;
(2)夏季水样中MW <1 kDa 和10 kDa <MW <0.45 μm 两部分DOM 为CH 的主要前体物;
(3)各分子量区间DOM 的3D-EEM 荧光光谱表征结果显示,CH 的主要前体物可能为类芳香性蛋白质、类微生物代谢产物等有机物而非类富里酸、类腐殖酸等有机物,来源可能是藻类的分泌物;
(4)模式化合物氯化试验结果显示:丙氨酸、天冬酰胺、天冬氨酸、半胱氨酸、组氨酸、苏氨酸、色氨酸、酪氨酸、蛋白质、DNA 等是CH 的主要前体物,CH 生成量均大于30 μg/mg C。
[1]Richardson S D,Postigo C. Drinking water disinfection by-products[M]. Springer Berlin Heidelberg:Emerging Organic Contaminants and Human Health,2012.
[2]Korshin G V. Development of Differential UV Spectroscopy for DBP Monitoring[M]. US:American Water Works Association,2002.
[3]Dąbrowska A,Nawrocki J. Controversies about the occurrence of chloral hydrate in drinking water[J]. Water research,2009,43(8):2201-2208.
[4]陆峰,向华.顶空气相色谱法测定水中三氯乙醛[J].净水技术,2006,25(6):71-73.
[5]Koudjonou B,LeBel G L,Dabeka L. Formation of halogenated acetaldehydes,and occurrence in Canadian drinking water[J].Chemosphere,2008,72(6):875-881.
[6]Pressman J G,Richardson S D,Speth T F,et al. Concentration,chlorination, and chemical analysis of drinking water for disinfection byproduct mixtures health effects research:US EPA's four lab study[J]. Environmental science & technology,2010,44(19):7184-7192.
[7]Simpson K L,Hayes K P. Drinking water disinfection by-products:an Australian perspective[J]. Water Research,1998,32(5):1522-1528.
[8]Wei J,Ye B,Wang W,et al. Spatial and temporal evaluations of disinfection by-products in drinking water distribution systems in Beijing,China[J]. Science of the Total Environment,2010,408(20):4600-4606.
[9]生活饮用水卫生标准,GB5749—2006[S].
[10]Guidelines for drinking-water quality:recommendations[S]. World Health Organization,2004.
[11]Gang D,Clevenger T E,Banerji S K. Relationship of chlorine decay and THMs formation to NOM size[J]. Journal of hazardous materials,2003,96(1):1-12.
[12]APHA,AWWA,WEF. Standard methods for the examination of water and wastewater[J]. American Public Health Association,Washington,DC,1998.
[13]Hua G,Reckhow D A. Characterization of disinfection byproduct precursors based on hydrophobicity and molecular size[J].Environmental science & technology,2007,41(9):3309-3315.
[14]Coble P G. Characterization of marine and terrestrial DOM in seawater using excitation-emission matrix spectroscopy[J]. Marine chemistry,1996,51(4):325-346.
[15]Leenheer J A,Croué J P. Peer reviewed:characterizing aquatic dissolved organic matter [J]. Environmental Science &Technology,2003,37(1):18-26.
[16]McKnight D M, Boyer E W, Westerhoff P K, et al.Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity[J].Limnology and Oceanography,2001,46(1):38-48.
[17]Chen W,Westerhoff P,Leenheer J A,et al. Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter[J]. Environmental science & technology,2003,37(24):5701-5710.
[18]Edzwald J K, Tobiason J E. Enhanced coagulation: US requirements and a broader view [J]. Water Science and Technology,1999,40(9):63-70.
[19]Matilainen A,Gjessing E T,Lahtinen T,et al. An overview of the methods used in the characterization of natural organic matter(NOM)in relation to drinking water treatment[J]. Chemosphere,2011,83(11):1431-1442.
[20]Panyapinyopol B,Marhaba T F,Kanokkantapong V,et al.Characterization of precursors to trihalomethanes formation in Bangkok source water[J]. Journal of Hazardous Materials,2005,120(1):229-236.
[21]Hureiki L,Croue J P,Legube B. Chlorination studies of free and combined amino acids[J]. Water Research,1994,28(12):2521-2531.
[22]Fang J,Yang X,Ma J,et al. Characterization of algal organic matter and formation of DBPs from chlor (am)ination[J]. Water Research,2010,44(20):5897-5906.
[23]Henderson R K,Baker A,Parsons S A,et al. Characterization of algogenic organic matter extracted from cyanobacteria,green algae and diatoms[J]. Water Research,2008,42(13):3435-3445.
[24] Chang H,Chen C,Wang G. Characteristics of C-,N-DBPs formation from nitrogen-enriched dissolved organic matter in raw water and treated wastewater effluent[J]. Water research,2013,47(8):2729-2741.
[25]Krasner S W. Contribution of wastewater to DBP formation[M].AWWA Research Foundation,2008:1-6.
[26]Liang S,Liu C,Song L. Soluble microbial products in membrane bioreactor operation: behaviors, characteristics, and fouling potential[J]. Water Research,2007,41(1):95-101.
[27]Trehy M L,Yost R A,Miles C J. Chlorination byproducts of amino acids in natural waters[J]. Environmental science & technology,1986,20(11):1117-1122.
[28]Chow C W K,van Leeuwen J A,Fabris R,et al. Optimised coagulation using aluminium sulfate for the removal of dissolved organic carbon[J]. Desalination,2009,245(1):120-134.
[29]Marttinen S K,Kettunen R H,Sormunen K M,et al. Screening of physical-chemical methods for removal of organic material,nitrogen and toxicity from low strength landfill leachates[J]. Chemosphere,2002,46(6):851-858.
猜你喜欢
天津医科大学学报(2021年1期)2021-12-05
纺织科学研究(2021年7期)2021-08-14
猪业科学(2018年4期)2018-05-19
现代检验医学杂志(2016年1期)2016-11-12
现代检验医学杂志(2016年5期)2016-08-20
国外医药(抗生素分册)(2016年4期)2016-07-12
浙江农业科学(2016年11期)2016-05-04
当代化工研究(2016年2期)2016-03-20
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年1期)2014-02-28
