
基于转录组数据的大菱鲆(Scophthalmus maximus)SNP标记开发及多态性分析*
2014-03-19 12:17黄智慧马爱军马得友王新安夏丹丹马本贺
海洋与湖沼 2014年6期
王 婷 黄智慧 马爱军① 马得友 王新安 夏丹丹 马本贺
(1. 中国水产科学研究院黄海水产研究所 青岛市海水鱼类种子工程与生物技术重点实验室 农业部海洋渔业可持续发展重点实验室 青岛 266071; 2. 大连海洋大学 大连 116023)
大菱鲆(Scophthalmus maximus)属于鲽形目、鲆科、菱鲆属, 在我国俗称“多宝鱼”。原产于欧洲大西洋东北部, 20世纪70年代, 英国首度成功开发了大菱鲆人工养殖技术, 于1992年引入我国, 目前已成为我国北方沿海工厂化养殖业的主导品种之一(雷霁霖等, 2002)。随着养殖规模的不断扩大, 集约化程度的不断提高, 大菱鲆疾病频发、养殖周期长、养殖成本上升等严重制约了我国大菱鲆的养殖业的可持续性发展(雷霁霖等, 2012)。因此, 对大菱鲆进行遗传改良、选育新品种已成为提高养殖效益和健康化养殖水平的当务之急。
大规模发掘大菱鲆分子标记, 是储备种质资源、构建性状相关遗传解析、开发分子遗传育种技术的基础工作。单核苷酸多态性标记(single nucleotide polymorphism, SNP)是美国学者Lander于1996年提出的第三代DNA遗传标记(Lander, 1996)。由于SNP数目多、覆盖密度大(Sachidanandamet al, 2001), 具有遗传稳定性和代表性(Collinset al, 1997), 便于高通量自动化检测等优点, 已经广泛应用在构建高密度遗传连锁图谱构建、关联分析、分子辅助育种、群体遗传系统、品种鉴定等方面, 并表现出良好的应用前景(Liuet al, 2004)。在水产动物中, SNP的开发和应用已经有若干报道, 如鲶鱼(Kucuktaset al, 2009)、栉孔扇贝(Jianget al, 2011)、蟹(Maet al, 2011)、虾夷扇贝(Liuet al, 2011)、对虾(吴莹莹等, 2013)等。在大菱鲆中也有部分开发和应用(刘庆明等, 2012; Navajaset al, 2012; Veraet al, 2013), 但数量还未达到构建高密度遗传连锁图谱的要求。
目前大规模SNP位点信息的获得主要是通过基因组测序, 伴随序列拼接比对而产生, 但是这种方法成本较高。对于没有基因组信息的非模式生物, 多利用已知的EST序列自行筛选, 但是EST序列与实际序列有一定差异, 所以这种方法前期准备工作比较艰巨, 开发效率比较低, 覆盖率不够广, 目的性不够强(Liuet al, 2006; 刘庆明等, 2012)。目前大菱鲆已知的EST序列只有15641个, 使得该物种SNP筛选工作难度更高; 随着第二代测序技术的快速发展, 高通量转录组测序成本大幅降低, 为解决这些难题提供了契机。通过新一代高通量测序技术, 能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本及基因序列, 并且可以准确检测出高度复杂的序列, 通过一系列对比识别大量的SNP位点(Zhouet al, 2010)。本研究首次利用大菱鲆雌雄高通量转录组测序信息, 去冗余后得到71107 contig,为大菱鲆的SNP标记大规模开发提供了丰富的序列资源和位点信息, 而且为进一步的QTL定位、性别控制等研究带来了极大便利。
迄今已经有很多种检测开发SNP多态性的技术方法, 如基于杂交的基因芯片技术(Gene chips)(Wanget al, 1998)、探针技术(TaqMan) (Martinsohnet al, 2009), 基于核酸构象的单链构象多态性(Singlestrand conformational polymorphism, SSCP) (Heet al,2008)等。虽然方法很多, 但是存在着开发量低、消耗时间多、操作步骤冗长、实验费用昂贵、适用范围有限等问题, 限制了SNP标记的利用和推广。高分辨率熔解曲线分析技术(High Resolution Melting, HRM),是近年来兴起的一种SNP检测的新手段, 可以迅速的检测出DNA片段中碱基的突变(Liet al, 2012); 因其操作简便快速, 结果准确效率高, 费用成本低, 并且实现了真正的闭管操作, 已受到广泛的关注(罗文龙等, 2011)。另外, HRM的小片段法特异性好, 避开了探针设计的误差和不对称PCR的复杂操作, 降低了开发成本, 提高了开发效率(Liewet al, 2004)。本研究在大菱鲆转录组Solexa高通量测序数据基础上,根据预测的SNP位点设计引物, 通过小片段HRM分型技术, 成功开发了大菱鲆EST-SNP标记21个, 为大菱鲆遗传图谱构建、性状相关QTL定位和分子遗传育种提供了候选标记资源。
1 材料与方法
1.1 实验材料
实验用大菱鲆来自本实验室在烟台天源水产国家级良种场所构建的家系(马爱军等, 2012), 共96尾鱼,样品详细资料见表1。DNA取样品尾鳍, 使用海洋动物组织基因组DNA提取试剂盒提取, 用超微量紫外分光光度计定量, 稀释至40ng/μL, –20°C保存备用。

表1 大菱鲆SNP标记开发样品信息Tab.1 Sample information of turbot for SNP in S. maximus
1.2 SNP位点的查找和小片段法引物设计
大菱鲆转录组Solexa高通量测序、contig的拼装和SNP位点的预测由国家人类基因组南方研究中心完成。根据高通量转录组测序数据结果, 查找覆盖度大于500的候选SNP位点, 共得到147个位点。
利用Primer5.0对每一个候选位点设计一组引物,要求引物长度20bp左右, GC含量为45%—65%, 产物长度小于80bp; 利用Oligo7.0分析引物参数, 不能有能值高的错配、二聚体和发卡结构。高低温内标为两段长度50bp的DNA片段, 提供独立于PCR产物的大约68°C和88°C的熔解曲线。分别为5-ATCGTG ATTTCTATAGTTATCTAAGTAGTTGGCATTAATAA TTTCATTTT-3’和5’-GCGGTCAGTCGGCCTAGCGG TAGCCAGCTGCGGCACTGCGTGACGCTCAG-3’及各自的反向互补序列(Seippet al, 2007)。引物和内标由上海生工生物工程技术服务有限公司合成。
1.3 PCR引物验证和HRM小片段法分型
以8个群体, 每个群体6个个体DNA等量混合作为模板DNA, 进行引物检测。PCR反应体系为10μL: 40 ng/μLDNA模板1μL, 2×ES Taq Master Mix 4.5μL, 上游引物(10μmol/L) 0.5μL; 下游引物(10μmol/L) 0.5μL,ddH2O 3.5μL。PCR反应程序为: 95°C预变性5min;94°C变性30 s, 退火温度(视具体引物Tm值而定)退火30s, 72°C延伸30s, 共35个循环; 最后72°C延伸7min, 4°C保存。用含Genefinder染料的2%琼脂糖凝胶电泳检测PCR扩增产物。
根据电泳结果, 选择目的条带清晰单一的位点进行高低温内标杂交并加入LC Green Plus染料。每个PCR反应体系中加入染料1.0μL, 高温内标和低温内标的正负向引物各加0.25μL, 95°C变性5min后,降温至4°C保存。
使用LightScanner仪器进行HRM分型, 要求以0.1°C/s的速度, 从56°C快速升温到97°C, 以1/s的密度采集荧光信号, 得到高分辨率溶解曲线。结束后用 LightScanner配套软件对采集曲线进行基因分型。
1.4 遗传多态性检测
SNP位点的分型结果采用POPGEN32 (version 1.32)软件处理, 纯合子分别记为AA和BB, 杂合子记为AB, 统计各位点的有效等位基因数(Effective number of alleles,Ne), 期望杂合度(Expected heterozygosity,He),观测杂合度(Observed heterozygosity,Ho), 统计各位点基因型分布及其频率和最小等位基因频率(Minor allele frequency, MAF)。用PIC_CALC(0.6)计算各位点多态信息含量(Polymorphism information content,PIC)。
2 实验结果
2.1 引物验证和小片段HRM分型
本课题组高通量测序系统大规模测序的结果共获得8642个SNP位点, 转换颠换比为1.3。在覆盖度大于500的SNP位点中, 根据两侧序列特点, 选取45个位点设计引物63组, 经过PCR体系条件优化, 设置最佳的Tm值, 利用2%的琼脂糖凝胶电泳进行检测,有21对引物产生大小符合预期的单一条带。其中部分SNP位点的详细信息见表2。对扩增出目的条带的位点, 用8个群体96个个体进行重新PCR, 变性, 经过小片段HRM (LightScanner)验证, 21个位点均能明显区分不同基因型。一共有11个颠换, 10个转换, 其中C/T和G/A最常见(图1)。
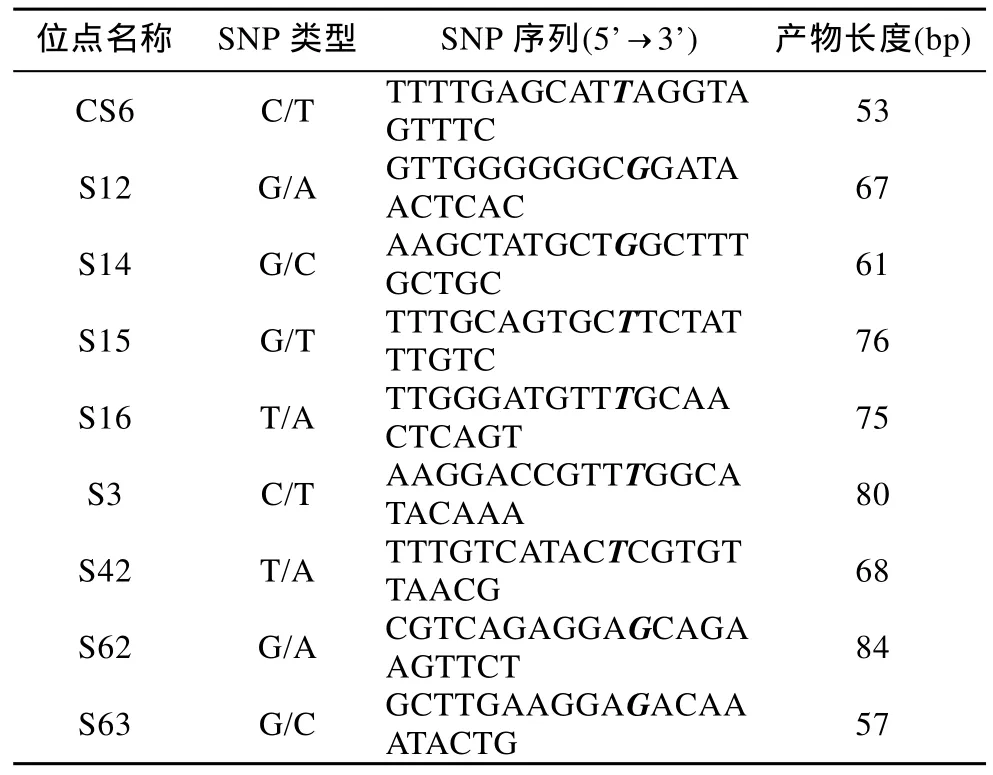
表2 部分SNP位点信息Tab.2 Detailed information of part of the SNP loci in S.maximus

图1 SNP突变类型分布Fig.1 Distribution of SNP variants
图2a所示为位点S3的小片段HRM分型图, 是转换, 其中红色曲线为纯合峰, 峰值较低,Tm约78.7°C,是T/T型; 蓝色曲线为纯合峰, 峰值较高,Tm值79.4°C, 是C/C型; 灰色为杂合峰, 是T/C型。图2b所示为位点S16的小片段HRM分型图, 是颠换, 其中红色曲线为纯合峰, 峰值较高,Tm约80.7°C, 是T/T型; 蓝色曲线为纯合峰, 峰值较低,Tm值80.1°C,是A/A型; 灰色为杂合峰, 是A/T型。
2.2 SNP位点多态性分析
所得的基因型数据利用POPGEN32(version 1.32)统计分析, 结果显示21个SNP位点均含有两个单倍体(表3); 观测杂合度Ho的分布范围为0.256—1.000,期望杂合度He的范围从为0.276—0.518。有效等位基因数Ne分布范围1.352—2.051。最小等位基因频率MAF分布范围0.0426—0.5000。多态信息含量分析显示21个位点的PIC值范围为0.226—0.446, 其中2个位点(CS5, S7)属于低度多态(PIC<0.25), 其余19个位点属于中度多态(0.25 利用NCBI的BlastP, 对成功筛查到的21个多态SNP位点所在的contig序列进行功能注释, 21个SNP位点所在的20个contig均有明确有效功能注释, 涉及细胞周期调控、细胞骨架、能量转换及RNA的加工修饰等(表4)。 利用ORF finder推测出开发阅读框, 在NCBI数据库中比对, 确保ORF选择的正确性, 并确定SNP位点在遗传密码子的位置, 与标准密码子比对发现CS1位于5’UTR区, S11、S15、S16、S42、S3、CS10位于3’UTR区; 而CS5、S7位于密码子的首位, CS9位于密码子第三位, 造成了编码氨基酸的改变, 为错义突变。其余11个位点都在密码子第三位, 为同义突变(表5)。 图2 SNP位点小片段HRM分型结果Fig.2 Genotyping result using HRM with small amplicon SNP标记的兴起, 极大地促进了遗传学和育种繁殖学的微观发展和深度发掘。然而, 开发和检测SNP是最关键的两个技术难点, 也限制了SNP标记在非模式生物(特别是水产动物)中的开发和利用。目前,SNP标记的大规模开发, 一般是利用基因组测序获得的序列结果进行对比推测, 这种途径比较昂贵(庄伟, 2010); 或者利用EST序列, 但是由于序列误差的存在, 增加了验证的难度(刘庆明等, 2012)。高通量转录组测序的推广很好地弥补了这两种途径的缺陷(周华等, 2012), 得到的大量EST序列既可以提供相对更准确的序列信息, 又降低了成本。本研究利用Solexa高通量测序技术获得大量EST-SNP位点, 经过生物信息学分析, 筛选出4314个覆盖度大于10的候选SNP位点, 明确了其基因功能。数量低于其它水产动物, 除物种差别, 另外可能的原因是筛查条件较为严格(Maet al, 2011; Veraet al, 2011)。 表3 大菱鲆21个SNP位点的遗传多态性Tab.3 Genetic variability at 21 SNPs in S. maximus 表4 20个序列预测蛋白的基因注释信息Tab.4 Gene annotation of 20 predicted proteins 表5 SNP位点的氨基酸突变Tab.5 Amino acid mutations of SNPs 由于转录组测序的样本是大菱鲆mRNA, 有一定的剪切和编辑, 本研究选取的45个SNP位点中有5个位点(11%)的PCR产物大于预期, 推测可能是由于内含子的存在而造成的。但是此数据远小于使用其它方法的比例(李纪勤等, 2013), 原因应是本研究使用的是小片段法HRM, 设计引物时, 使目的片段尽可能地小, 有效避免了内含子的出现。另外有4个位点,有可观的多态性, 但是没有准确的分型结果, 因为产物溶解峰与高温内标溶解峰有部分重叠。要解决这个问题, 需要溶解温度更高的高温内标, 或者重新设计引物以期得到溶解温度更低的目的片段。 本研究采用高分辨率溶解曲线(HRM)技术, 并探索了小片段法分型技术, 通过该方法可以有效避免在同一个目标产物中存在2个以上SNP位点的可能性, 减少了内含子的影响, 从而提高分型效率, 大幅度降低了SNP标记开发的成本。本研究对45对SNP位点进行引物设计、验证、基因分型、多态性分析最终获得21个SNP标记可用于大菱鲆的遗传和育种研究, 成功率达到46.7%, 远远高于非标记探针法(刘庆明等, 2012; 吴莹莹等, 2013)。 对获得的21个SNP标记, 在群体中进行遗传多态性验证, 多态信息(PIC)含量分析显示所有位点均具有多态性, 19个为中等多态。观测杂合度的分布范围为0.256到1.000, 期望杂合度的平均值为0.451,由于SNP标记为双等位形式, 每个位点所携带的多态信息较少, 因此略小于大菱鲆的其它分子标记(Houet al, 2011; Veraet al, 2013)。另外有两个偏离了Hardy-Weinberg平衡, 说明样本群体可能有过迁徙漂变等。 利用EST数据库开发SNP标记, 有一个显著的优点, 即标记位点来自编码序列(Coding Sequences,CDS), 与功能基因直接相关, 为进一步的功能基因研究提供依据。本研究获得3个非同义cSNP (nonsynonymous cSNP), 即碱基序列的改变可使以其为蓝本翻译的蛋白质序列发生改变, 从而影响了蛋白质的功能(刘越等, 2008)。另外, 有7个位于基因的UTL区,可能与基因的表达调控相关(Veraet al, 2011, 2013),后续基因功能表达途径等跟进研究中。 总之, 对于大菱鲆这种没有基因组数据的非模式生物, 应用高通量转录组测序结果, 对比EST序列, 使用小片段HRM技术, 可以实现高效率大规模SNP标记的开发, 而且开发位点与功能基因表达途径直接相关, 为进一步的遗传连锁图谱构建、经济性状的QTL定位以及分子标记辅助育种提供更多的标记基础。 致谢 中国科学院海洋研究所张国范研究员课题组在实验仪器方面提供了帮助, 谨致谢忱。 马爱军, 黄智慧, 王新安等, 2012. 大菱鲆(Scophthalmus maximus)耐高温品系选育及耐温性能评估. 海洋与湖沼,43(4): 797—804 庄 伟, 2010. 半滑舌鳎(Cynoglossus semilaevis)基因组SNP标记的开发与检测. 青岛: 中国海洋大学硕士学位论文,1—57 刘 越, 吕社民, 2008. 单核苷酸多态性影响基因功能的机制.生命的化学, 28(2): 214—216 刘庆明, 李 猛, 马爱军等, 2012. 应用高分辨率熔解曲线(HRM)开发大菱鲆(Scophthalmus maximus)SNP标记的研究. 海洋与湖沼, 43(6): 1141—1148 李纪勤, 包振民, 李 玲等, 2013. 栉孔扇贝EST-SNP标记开发及多态性分析. 中国海洋大学学报(自然科学版), 43(1):56—63 吴莹莹, 孟宪红, 孔 杰等, 2013. 非标记探针HRM法在中国对虾EST-SNP筛选中的应用. 渔业科学进展, 34(1):111—118 罗文龙, 郭 涛, 王 慧等, 2011. HRM及其在植物育种中的应用展望. 中国农学通报, 27(3): 10—14 周 华, 张 新, 刘腾云等, 2012. 高通量转录组测序的数据分析与基因发掘. 江西科学, 30(5): 607—611 雷霁霖, 王秉新, 2002. 大菱鲆“温室大棚+深井海水”工厂化养殖模式. 海洋水产研究, 23(4): 1—7 雷霁霖, 刘新富, 关长涛, 2012. 中国大菱鲆养殖20年成就和展望——庆祝大菱鲆引进中国20周年. 渔业科学进展,33(4): 123—130 Collins F S, Guyer M S, Chakravarti A, 1997. Variations on a theme: cataloging human DNA sequence variation. Science,278(5343): 1580—1581 He F, Wen H S, Dong S Let al, 2008. Identification of single nucleotide polymorphism cytochrome P450-c19a and its relation to reproductive traits in Japanese flounder(Paralichthys olivaceus). Aquaculture, 279(1): 177—181 Hou S, Ma A, Wang X Aet al, 2011. Isolation and characterization of 45 Polymorphie microsatellite loci of turbot (Scophthalmus maximus) and cross-species amplification. Chinese Journal of Oceanology and Limnology,29: 311—316 Jiang G, Li J, Li Let al, 2011. Development of 44 gene-based SNP markers in Zhikong scallop,Chlamys farreri.Conservation Genetics Resources, 3(4): 659—663 Kucuktas H, Wang S, Li Pet al, 2009. Construction of genetic linkage maps and comparative genome analysis of catfish using gene-associated markers. Genetics, 181(4):1649—1660 Lander E S, 1996. The new genomics: global views of biology.Science, 274(5287): 536—539 Li F, Niu B, Huang Yet al, 2012. Application of high-resolution DNA melting for genotyping in lepidopteran non-model species:Ostrinia furnacalis(Crambidae). PloS One, 7(1):e29664 Liew M, Pryor R, Palais Ret al, 2004. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clinical Chemistry, 50(7): 1156—1164 Liu W, Li H, Bao Xet al, 2011. The first set of EST-derived single nucleotide polymorphism markers for Japanese scallop,Patinopecten yessoensis. Journal of the World Aquaculture Society, 42(3): 456—461 Liu Y G, Liu L X, Lei Z Wet al, 2006. Identification of polymorphic microsatellite markers from RAPD product in turbot (Scophthalmus maximus) and a test of cross-species amplification. Molecular Ecology Notes, 6(3): 867—869 Liu Z J, Cordes J F, 2004. DNA marker technologies and their applications in aquaculture genetics. Aquaculture, 238(1):1—37 Ma H, Ma Q, Ma Cet al, 2011. Isolation and characterization of gene-derived single nucleotide polymorphism (SNP)markers inScylla paramamosain. Biochemical Systematics and Ecology, 39(4): 419—424 Martinsohn J T, Ogden R, 2009. FishPopTrace-Developing SNP-based population genetic assignment methods to investigate illegal fishing. Forensic Science International:Genetics Supplement Series, 2(1): 294—296 Navajas Pérez R, Robles F, Molina Luzón M Jet al, 2012.Exploitation of a turbot (Scophthalmus maximusL.)immune-related expressed sequence tag (EST) database for microsatellite screening and validation. Molecular Ecology Resources, 12(4): 706—716 Sachidanandam R, Weissman D, Schmidt S Cet al, 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature, 409(6822):928—933 Seipp M T, Durtschi J D, Liew M Aet al, 2007. Unlabeled oligonucleotides as internal temperature controls for genotyping by amplicon melting. The Journal of Molecular Diagnostics, 9(3): 284—289 Vera M, álvarez-Dios J A, Millán Aet al, 2011. Validation of single nucleotide polymorphism (SNP) markers from an immune Expressed Sequence Tag (EST) turbot,Scophthalmus maximus,database. Aquaculture, 313(1): 31—41 Vera M, Alvarez-Dios J, Fernandez Cet al, 2013. Development and validation of single nucleotide polymorphisms (SNPs)markers from two transcriptome 454-runs of turbot(Scophthalmus maximus) using high-throughput genotyping. International Journal of Molecular Sciences, 14(3):5694—5711 Wang D G, Fan J, Siao Cet al, 1998. Large-scale identification,mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science, 280(5366): 1077—1082 Zhou X, Ren L, Li Yet al, 2010. The next-generation sequencing technology: a technology review and future perspective.Science China Life Sciences, 53(1): 44—572.3 SNP位点序列的功能分析

3 讨论



猜你喜欢
黑龙江水产(2022年6期)2022-12-03
透析与人工器官(2020年1期)2020-11-16
基层中医药(2020年5期)2020-09-11
铁道通信信号(2019年8期)2019-10-10
中国水产(2019年3期)2019-03-25
基层中医药(2018年5期)2018-08-31
中国发展观察(2017年8期)2017-04-26
浙江农业学报(2016年7期)2016-06-15
制造技术与机床(2015年10期)2015-04-09
中国当代医药(2015年33期)2015-03-01
