载利福喷丁/聚乳酸-聚乙醇酸共聚物微球制备及体外释放
2014-03-14 03:53胡运玖李吉东李玉宝蒋电明
脊柱外科杂志 2014年6期
邬 均,胡运玖,左 奕,李吉东,李玉宝,蒋电明
微球制剂是目前国内外药学领域研究的热点,用生物可降解材料作为药物载体,通过注射或植入给药,在局部缓慢释放药物,减少给药次数,并能维持局部较长时间的高药物浓度。目前研究和应用最广泛的微球载体材料是聚乳酸-聚乙醇酸共聚物[Poly(lactic-co-glycolic acid), PLGA]。PLGA具有良好的生物相容性及生物可降解性,无毒、无刺激性,被美国食品与药物管理局批准广泛运用于载药系统研制[1]。利福喷丁与利福平同属利福霉素家族,两者抗菌谱及作用机制相似,同被美国Medical Letter治疗指南(2007年)列为一线抗结核药物,但利福喷丁抗结核杆菌作用明显高于利福平,与异烟肼联合用药的作用更强,且利福喷丁半衰期更长、不良反应更轻微,与利福平相比具有高效、长效、低毒的特点[2]。
本研究通过乳化-溶剂挥发法制备载利福喷丁/PLGA微球,通过形貌、粒径及分布、载药量及包封率等评价载药微球质量,并对载药微球体外释放特性进行研究,以期获得一种有效的抗结核制剂。
1 材料及方法
1.1 材料及试剂
PLGA(50∶50,平均分子量45 000,山东岱罡生物技术公司);利福喷丁原药(分装,德国Ruibio公司);Viskase透析袋(截留相对分子质量3 500,美国联合碳化公司)。结核分枝杆菌标准菌毒株H37Rv(重庆市结核基因诊断中心实验室)。二氯甲烷、聚乙烯醇、甲醇(上海阿拉丁试剂公司)均为分析纯级。
1.2 仪器
JJ-1型精密增力电动搅拌器(常州国华电器有限公司);FD-1A-50型冷冻干燥机(北京博医康实验仪器有限公司);TE2000-U型倒置相差显微镜(日本Nikon公司);JEOL-6500LV型扫描电镜(日本电子公司);JL-6000型激光粒度仪(四川成都精新粉体测试设备有限公司);754型紫外分光光度计(上海光谱仪器有限公司)。
1.3 实验方法
1.3.1 载利福喷丁微球/PLGA制备
采用乳化-溶剂挥发法(O/W法)[1,3]制备载利福喷丁/PLGA微球。准确称取1 g PLGA溶于10 mL二氯甲烷溶液,超声溶解完全后,加入400 mg利福喷丁原药,溶解混匀形成油相。将油液缓慢滴加至搅拌着的含有2%PVA的100 mL水溶液,高速搅拌乳化30 min,形成O/W乳液。搅拌速率下调为约250 r/min,持续均速搅拌至二氯甲烷挥发完全。静置1 h后离心收集微球,用去离子水反复洗涤、离心3次。将所得微球转移至西林瓶中,置于低温冷冻干燥机中冻干。-20℃密封保存备检。
1.3.2 形貌观察
取少量载药微球分散于蒸馏水中,滴加至载玻片,在光学显微镜下观察微球形貌。另取适量载利福喷丁/PLGA微球用蒸馏水分散于导电胶上,蒸发干燥后镀金,扫描电镜下观察微球形貌。
1.3.3 粒径及其分布
将适量载利福喷丁/PLGA微球加入激光粒度仪,同时滴加2滴分散剂,激光粒度仪分析测定微球粒径。微球平均粒径以中值粒径D50表示,采用跨距评价粒径分布情况,跨距=(D90-D10)/D50。
1.3.4 载药量及包封率测定
配制不同浓度梯度的利福喷丁标准溶液,并在475 nm波长处紫外分光光度计测定其吸光度,绘制利福喷丁紫外吸收标准曲线。
准确称取载利福喷丁/PLGA微球10 mg,加入10 mL甲醇溶剂,(37±1)℃恒温震荡至载药微球完全变白,离心,取上层液体2.5 mL,甲醇溶液稀释至25 mL。采用紫外分光光度计测量其吸光度值,并采用标准曲线计算利福喷丁浓度。根据以下公式计算载利福喷丁/PLGA微球的载药量及包封率:载药量(%)=微球中所含利福喷丁质量/微球质量×100%;包封率(%)=微球中所含利福喷丁质量/投入福喷丁质量×100%
1.3.5 利福喷丁抗结核分枝杆菌最低抑菌浓度
结核分枝杆菌标准菌毒株H37Rv接种于改良罗氏培养基,37℃、5% CO2孵箱中培养4周,选取单个较大菌落,配制成结核分枝杆菌菌悬液(1 mg/mL),过滤后菌悬液分装于无菌菌种保存管,-70℃保存备用[4]。
将利福喷丁溶解于无菌去离子水,取不同量加入改良罗氏培养基,使培养基含有利福喷丁浓度分别为32 μg/mL、16 μg/mL、8 μg/mL、4 μg/mL、2 μg/mL、1 μg/mL、0. 500 μg/mL、0.250 μg/mL、0.125 μg/mL和0 μg/mL。85℃凝固灭菌1 h后取出,无菌试验合格后备用。
将冻藏于-70℃的菌悬液室温溶解,稀释至0.01 mg/mL。用微量加样器吸取0.1 mL分别接种于上述培养基上。5% CO2孵箱中37℃培养4周观察结果,完全无结核分枝杆菌生长的利福喷丁最低浓度培养基为该药物的最低抑菌浓度(minimal inhibitory concentration, MIC)。
1.3.6 体外释放实验
精确称取载利福喷丁/PLGA微球10 mg,装入透析袋内,扎紧并完全浸入10 mL PBS缓冲溶液(0.2 mol/L,pH=7.4)。37.1℃恒温振荡箱中,以100 r/min震荡。于预设时间点取出PBS缓冲液,并加入10 mL新鲜PBS缓冲液。离心并取上层清液,紫外分光光度计测定其吸光度,采用标准曲线计算利福喷丁浓度。每份样品重复试验3次。根据标准曲线方程计算利福喷丁累积释放量及药物累积释放百分率,绘制时间-累计释放百分率曲线。
2 结 果
2.1 微球形貌观察
大体观察,微球外观呈橘红色细粉状,无聚集成团。光镜下微球呈橘黄色球状,微球分散好,无粘连(见图1a)。扫描电镜下载药微球呈圆球状,均匀分散,无粘连成团,大小较均一(见图1b);放大后观察,微球表面致密、光滑(见图1c)。
2.2 微球粒径分布
载利福喷丁/PLGA微球粒径分布情况采用激光粒度仪分析,结果如图2所示。载药微球体积平均粒径D50为25.49 μm,跨距为1.59,粒径分布范围较窄,表明所制备的微球粒径均匀。
2.3 微球载药量及包封率测定
载利福喷丁/PLGA微球的载药量及包封率分别为21.37%±0.16%和 74.79%±2.71%。
2.4 利福喷丁抗结核分枝杆菌最低抑菌浓度
观察发现,在2 μg/mL及更高浓度的培养基中完全无结核分枝杆菌生长,而在更低浓度的培养基中可见数量不等的菌落。因此,利福喷丁抗结核分枝杆菌的最低抑菌浓度为2 μg/mL。
2.5 载利福喷丁/PLGA微球体外释放实验
载利福喷丁/PLGA微球体外释放实验结果如表1所示。根据实验结果绘制时间-累计释放百分率曲线,如图3所示。

a: 光镜 (×40) b: 扫描电镜(×50) c: 扫描电镜 (×1 000)
a: Light microscope (×40) b: Scanning electron microscope (×50) c: Scanning electron microscope (×1 000)
图1光镜及扫描电镜观察微球形貌
Fig.1Morphology of rifapentine-loaded PLGA microspheres
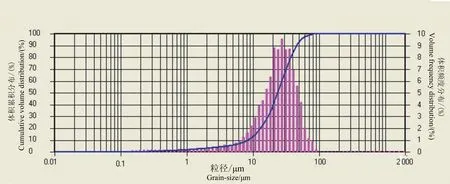
图2载利福喷丁/PLGA微球粒径分布
Fig.2Grain-size distribution of rifapentine-loaded PLGA microspheres
由表1和图3可看出,利福喷丁原药仅2 d释药量就为94.25%±3.74%,第3天释药量低于最低抑菌浓度。载利福喷丁/PLGA微球体外释放存在明显的两相性,即突释相和缓释相。在最初12 h突释相内利福喷丁释放量占载药微球中药物含量的37.08%±1.68%,12 h后载药微球释药速度减慢,35 d后体外累积释放量达80.67%±0.97%。载利福喷丁/PLGA微球在实验周期(0~35 d)内任一时间点的药物释放浓度均高于利福喷丁抗结核分枝杆菌的最低抑菌浓度。由此可知,载利福喷丁/PLGA微球具有良好的缓释性能,利福喷丁可从载药微球中持续释放。
表1利福喷丁及载利福喷丁/PLGA微球体外释放
Tab.1In vitro release of pure rifapentine and rifapentine-loaded PLGA microspheres
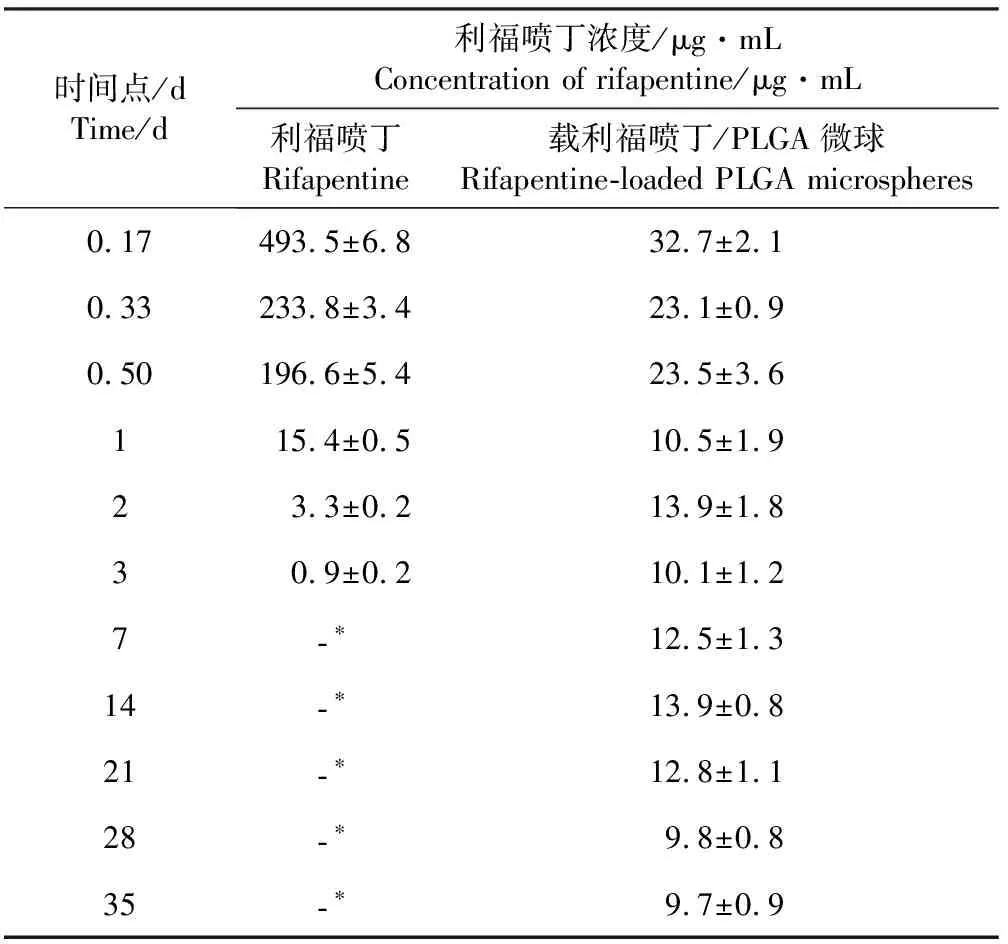
时间点/dTime/d利福喷丁浓度/μg·mLConcentration of rifapentine/μg·mL利福喷丁Rifapentine载利福喷丁/PLGA微球Rifapentine-loaded PLGA microspheres0.17493.5±6.832.7±2.10.33233.8±3.423.1±0.90.50196.6±5.423.5±3.6115.4±0.510.5±1.923.3±0.213.9±1.830.9±0.210.1±1.27-∗12.5±1.314-∗13.9±0.821-∗12.8±1.128-∗9.8±0.835-∗9.7±0.9
注:*利福喷丁原药在第3天释药量低于其抗结核分枝杆菌的最低抑菌浓度,故未继续测量
Note: *Release dosage of pure rifapentine below its minimum inhibitory concentration against Mycobacterium tuberculosis in the third day. So the meansurenent did not continue
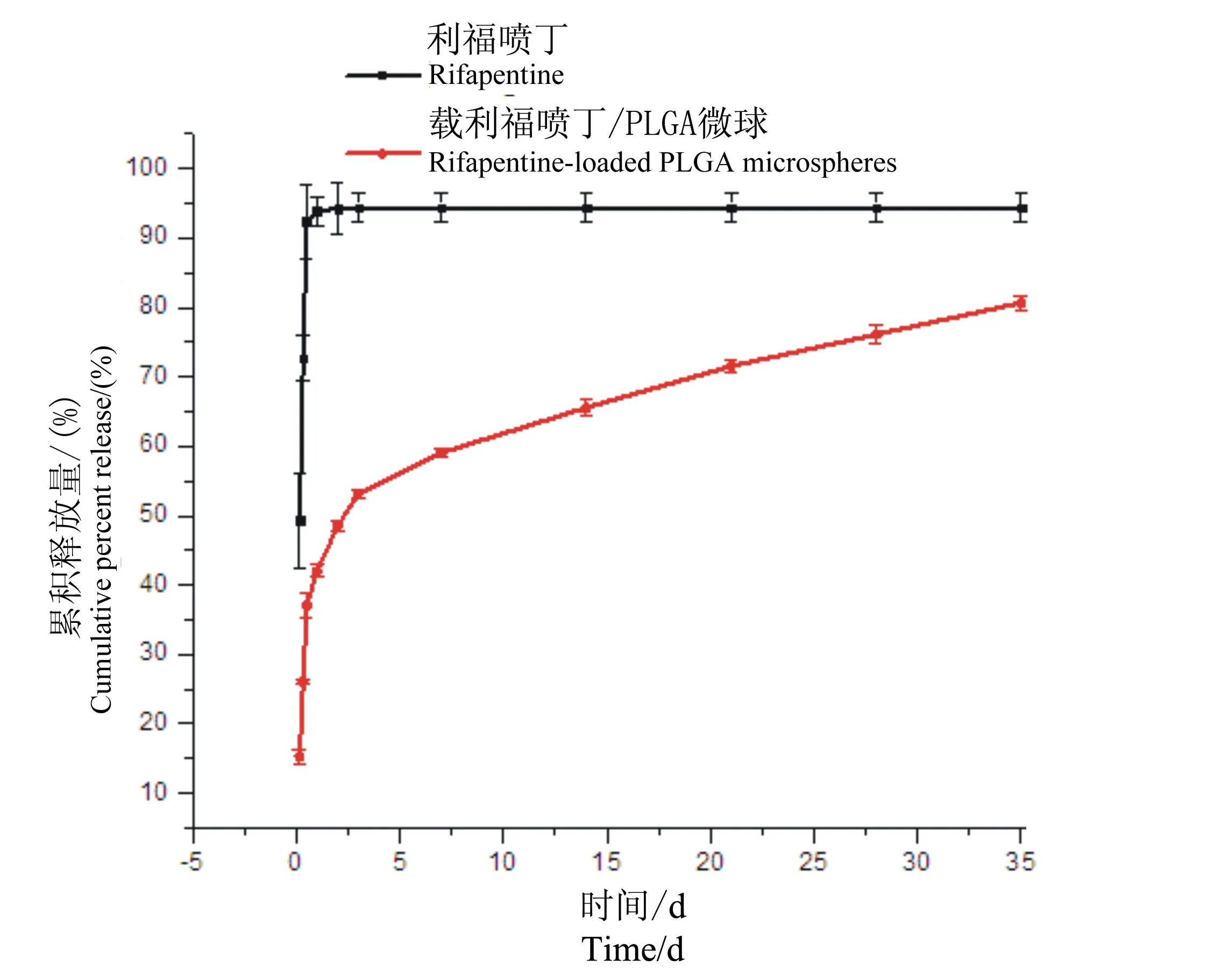
图3利福喷丁原药及载利福喷丁/PLGA微球体外释放曲线
Fig.3In vitro release profile of pure rifapentine and rifapentine-loaded PLGA microspheres
3 讨 论
骨与关节结核是临床上常见的肺外结核,约50%累积脊柱,脊柱结核占全部结核的3%~5%。脊柱结核易导致脊柱失稳、后凸畸形及脊髓神经功能障碍,约10%脊柱结核患者并发截瘫,给患者健康和生活质量造成巨大的影响[5]。脊柱结核治疗主要包括全身治疗(休息、营养、支持治疗)、抗结核药物化疗及外科手术治疗[6]。药物化疗是脊柱结核治疗的基础,有效杀灭结核杆菌是治愈脊柱结核根本措施,化疗应严格遵循早期、规律、全程、联合、适量的原则,具体治疗方案可参考我国骨与关节结核标准化疗方案和短程化疗方案[7-8]。脊柱结核外科治疗适用于脊柱稳定性破坏、严重或进行性后凸畸形、脊髓受压伴神经功能障碍,其目的是彻底清除病变组织,提高组织修复能力,解除脊髓神经压迫,矫正畸形,重建脊柱稳定性。结核病灶的彻底清除是各种术式的基础,除了彻底清除死骨、干酪样坏死物质、脓肿、结核肉芽、被破坏的椎间盘等组织,应特别注意彻底刮除病灶周围硬化骨。硬化骨不仅不利于局部植骨融合,还是抗结核药物进入病灶的巨大屏障,而且硬化骨中含有较多的结核杆菌,如果不彻底清除,可能成为术后复发的根源[9]。但传统的脊柱结核病灶清除术很难将病灶彻底清除,临床医生常规在病灶处空腔内喷洒适量普通剂型的抗结核药物,但普通剂型抗结核药物容易被机体代谢,术后复发率高(1.28%~25%)[10]。因此,研发出一种能在病灶局部缓慢释放,进而发挥持续杀灭结核杆菌的新型制剂成为亟待解决的问题。
微球制剂通过载体向患部持续和高浓度地释放药物以达到临床治疗目的,有利于提高药物疗效、降低毒副作用,对于提高临床用药水平来说具有重大意义[11]。微球制剂的载体是高分子材料,按降解性分为不可降解生物材料(聚丙烯、乙基纤维素等)和可降解生物材料,后者包括天然高分子材料(白蛋白、明胶、壳聚糖、海藻酸盐等)和合成高分子材料(聚乳酸、PLGA、聚己内酯等)[12]。生物降解性合成高分子材料无毒性,生物相容性好,可完全生物吸收或降解,不需二次手术取出,并能通过调整聚合物的组成或分子量的大小调整降解速度和药物释放速度,以满足临床需求。PLGA是目前研究最为广泛的可降解性合成高分子微球载体材料,截止到目前,已经有8种PLGA可注射微球型剂被美国食品与药物管理局批准用于临床[1,11,13]。
利福喷丁为半合成利福霉素类抗生素,通过与依赖DNA的RNA多聚酶的亚单位牢固结合,抑制细菌RNA的合成,在转录水平发挥杀菌作用[14]。利福喷丁与利福平同为一线抗结核药物,其最低抑菌浓度为0.12 ~0.25 mg/L,比利福平强2~10倍,且利福喷丁半衰期为18 h,明显长于利福平(3~5 h)。与利福平相比,利福喷丁能维持局部更长时间的有效抑菌浓度和更加高效的抑菌效果。
本研究采用PLGA为微球载体材料,采用利福喷丁作为模型药物,通过乳化-溶剂挥发法制备载利福喷丁/PLGA微球。形貌观察及粒径分析结果表明微球呈橘黄色,分散性好,无粘连现象,微球成球形态良好,微球表面致密、光滑,无凹凸现象,粒径分布较均匀。
载药量和包封率是衡量微球制备工艺的个重要指标。载药量是微球中所含目标药物的质量百分数,提高的载药量不仅可以节约所需载体材料量,还可以提高组织对目标药物的摄取效率。包封率是微球实际载药量与理论载药量的比值,包封率越高,在制备过程中所浪费的目标药物就越少。因此,载药量和包封率越高越好。影响微球载药量及包封率的主要因素有PLGA的组成、目标药物的理化性质、各溶剂相用量、微球制备方法和参数[15]。本研究所制备的利福喷丁/PLGA微球的载药量及包封率分别为21.37%±0.16%和 74.79%±2.71%,高于Dutt等[16]报道的12%~14%载药量。
载药微球的药物释放动力学特性是评价微球质量的内在指标,是微球质量控制的重要手段。随着研究的不断深入,对微球制剂的药物释放有了更高层次的要求,即从仅要求平稳的血药浓度提高到要求释药速度能使靶位在治疗所需的时间内维持合适的药物浓度。本研究制备的载利福喷丁/PLGA微球体外药物释放可以分为突释阶段和缓释阶段。在突释阶段,载药微球释放利福喷丁速度较快,药物释放量约占微球药物含量的40%,可能是表层吸附药物的释放。随后药物释放速度减慢,呈现持续而缓慢的药物释放,每个时间点的利福喷丁释放浓度均大于其最低抑菌浓度,35 d后体外累积释放量达80%。这种2阶段药物释放特性对脊柱结核的治疗有重要作用,突释阶段的药物快速释放可使病灶快速达到有效的抑菌浓度,杀灭术后病灶残留的结核杆菌,而后期稳定的药物释放可使局部得以维持较长时间有效杀菌药物浓度,防止结核复发。
采用乳化溶剂挥发法制备的载利福喷丁/PLGA微球,载药微球成球形良好,平均粒径为25.49 μm,具有较高的载药量及包封率,体外释放结果显示其具有稳定的缓释作用,35 d后利福喷丁释放浓度仍是利福喷丁体外抗结核分枝杆菌最低抑菌浓度的5倍,可作为一种较为理想的抗脊柱结核药物释放系统。尚需进一步对载药微球生物相容性、体内降解及体内药代动力学等进行研究。
参考文献
[1] Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices[J]. Biomaterials, 2000, 21(23):2475-2490.
[2] 徐学昌. 利福喷丁与利福平在肺结核治疗中的药效比较及安全性评价[J].海峡药学,2012,24(4):85-86.
[3] Song Cunxian, Sun Hongfan, Yang Jin, et al. Cytochalasin B loaded nano/microparticles: preparation and in vitro drug release evaluation[J]. 中国生物医学工程学报(英文版), 2000, 9(1):1-9.
[4] 黎友伦, 陈保文, 沈小兵, 等. 结核分枝杆菌接种菌量对异烟肼和利福平药敏试验的影响[J]. 中国防痨杂志, 2006, 28(5):288-291.
[5] 胥少汀, 葛宝丰, 徐印坎. 实用骨科学[M].第3版, 北京:人民军医出版社, 2006:1278.
[6] Jutte PC, Van Loenhout-Rooyackers JH. Routine surgery in addition to chemotherapy for treating spinal tuberculosis[J]. Cochrane Database Syst Rev, 2013, 5(1):1-38.
[7] Van Loenhout-Rooyackers JH, Verbeek AL, Jutte PC. Chemotherapeutic treatment for spinal tuberculosis[J]. Int J Tuberc Lung Dis, 2002, 6(3):259-265.
[8] 金大地. 脊柱结核治疗若干问题探讨[J]. 脊柱外科杂志, 2005, 3(3):186-188.
[9] 王自立. 脊柱结核手术治疗的相关问题探讨——脊柱结核的病灶清除与融合固定问题[J].中国脊柱脊髓杂志, 2006, 16(12):888- 889.
[10] Garg RK, Raut T, Malhotra HS, et al. Evaluation of prognostic factors in medically treated patients of spinal tuberculosis. Rheumatol Int, 2013,33(12):3009-3015.
[11] Freiberg S, Zhu XX. Polymer microspheres for controlled drug release[J]. Int J Pharm, 2004, 282(1-2):1-18.
[12] Edlund U, Albertsson AC. Degradable polymer microspheres for controlled drug delivery [J]. Adv Poly Sci, 2002 (157):67-112.
[13] 闫丽萍, 王海学, 彭健. 微球制剂的临床前药理毒理研究和评价[J]. 药物评价研究, 2010, 33(5):332-334.
[14] Katzung BG. Basic & Clinical Pharmacology[M]. 8th ed, San Francisco: McGraw -Hill, 2001.
[15] 孙美丽, 班俊峰, 黄思玉, 等.PLGA微球载药量和包封率的影响因素及控制[J].广东药学院学报, 2011, 27(6):643-648.
[16] Dutt M, Khuller GK. Chemotherapy of Mycobacterium tuberculosis infections in mice with a combination of isoniazid and rifampicin entrapped in Poly (DL-lactide-co-glycolide) microparticles[J]. J Antimicrob Chemother, 2001, 47(6):829-835.
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
潍坊学院学报(2020年6期)2020-11-22
海峡科技与产业(2016年3期)2016-05-17
中国卫生标准管理(2015年25期)2016-01-14
小说月刊(2015年6期)2015-12-16
大连工业大学学报(2015年4期)2015-12-11
中国当代医药(2015年32期)2015-03-01
中国当代医药(2015年29期)2015-03-01
中国医药导报(2015年26期)2015-02-28