
RP-HPLC法测定葛藤分散片中葛根素与大豆苷的含量
2014-03-07 10:26董宏伟刘万卉
药学研究 2014年2期
董宏伟,刘万卉
(烟台大学药学院,山东烟台264005)
RP-HPLC法测定葛藤分散片中葛根素与大豆苷的含量
董宏伟,刘万卉
(烟台大学药学院,山东烟台264005)
目的建立同时测定葛藤分散片中葛根素与大豆苷含量的反相高效液相色谱法。方法采用Agilent Zarbax Eclipse C18色谱柱(4.6 mm×150 mm,5μm),流动相为甲醇-水(20∶80,V/V),流速1.0 mL·min-1,检测波长250 nm,柱温35℃,进样量10μL。结果葛根素和大豆苷在各自的线性范围内(葛根素:9.04~72.32μg· mL-1;大豆苷:9.12~72.96μg·mL-1),峰面积与浓度呈良好的线性关系(r值均大于0.999)。葛根素和大豆苷的最低定量限分别为0.45和0.46μg·mL-1,加样回收率分别为100.2%和101.7%,R SD分别为0.7%和0.9%(n=9)。结论本方法简便、准确、灵敏、重现性好,专属性强,可用于葛藤分散片中葛根素和大豆苷的质量控制。
葛藤分散片;葛根素;大豆苷;高效液相色谱法
葛藤与葛根的总黄酮含量相近,葛藤中大豆苷含量较葛根素高。随着市场需求的扩大,葛根的过度挖掘对环境造成了不可逆转的危害,因此开展葛藤总黄酮的开发研究不仅具有较大的经济效益,更有保护资源和水土流失的环保及社会意义。
葛藤为豆科植物野葛Pueraria Lobata(Wild)ohwi的地上部分干燥茎,其药用价值近年来受到国内外学者的关注,其主要含有大豆苷元(Daidzein)、葛根素(Puerarin)等异黄酮成分。葛根素有扩张心脑血管,降低心肌氧耗,改善心肌收缩功能和促进微循环的作用[1],对葛根素的测定方法主要有紫外可见分光光度法、薄层色谱法、高效液相色谱法和毛细管电泳法等[2]。本试验采用RP-HPLC以等度洗脱的方法,同时测定葛藤分散片中葛根素和大豆苷的含量,样品中葛根素和大豆苷与其他物质的分离度较好。
1 仪器与试剂
1.1 仪器 Agilent1100液相色谱仪,单元泵,VWD检测器,Agilent色谱工作站,7725i手动进样阀;SK2200H超声清洗仪(上海科导超声仪器有限公司);十万分之一电子分析天平(奥豪斯仪器上海有限公司)。
1.2 试剂 葛根素对照品(批号:110752-200511)、大豆苷对照品(批号:111738-200501);皆由中国药品生物制品检定所提供;葛藤分散片(批号:20070110,20070112,20070121);阴性葛藤分散片,由药学院药剂室提供;甲醇、乙腈(色谱纯,Burdick&Jackson);水为二次重蒸水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件 Agilent Zarbax Eclipse C18柱(4.6 mm×150 mm,5μm),柱温:35℃,流动相为甲醇-水(20∶80,V/V),流速1.0 mL·min-1,检测波长:250 nm,进样量:10μL。
在该色谱条件下葛根素、大豆苷的保留时间分别为8.9、15.5 min,葛藤分散片的色谱见图1。
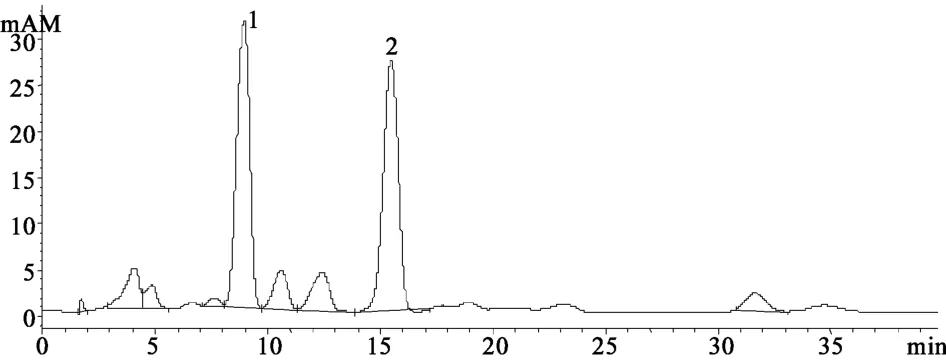
图1 葛藤分散片的HPLC色谱图 1.葛根素;2.大豆苷
2.2 供试品溶液的制备 取本品10片研碎后取粉末约0.5 g,精密称定,置锥形瓶中加甲醇50 mL超声萃取30 min,放冷,用甲醇定容于100 mL量瓶中,取过0.45μm微孔滤膜的续滤液1 mL,置10 mL量瓶中加甲醇稀释至刻度,摇匀,作为供试品溶液[3~5]。
2.3 对照品溶液的制备 精密称取经五氧化二磷干燥过的葛根素、大豆苷对照品各约10 mg,精密称定,置50 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为对照品溶液。
2.4 线性关系考察 精密称取葛根素、大豆苷对照品适量,加甲醇溶解并稀释制成浓度分别为9.04、18.08、27.12、36.16、72.32μg·mL-1的葛根素系列标准溶液和浓度分别为9.12、18.24、27.36、36.48、72.96μg·mL-1大豆苷的系列标准溶液,照“2.1”项下色谱条件方法测定峰面积,记录色谱图[6,7],以峰面积Y对浓度X做线性回归,葛根素回归方程为:Y=47.944X+25.793,r=0.999 3,大豆苷:Y=42.33X+21.73,r=0.999 1,表明葛根素浓度在9.04~72.32μg·mL-1、大豆苷浓度在9.12~72.96μg·mL-1范围内,峰面积与浓度呈良好的线性关系。
2.5 定量限和检测限 分别精密称取葛根素和大豆苷对照品适量,加甲醇溶解并逐步稀释制成一定浓度的溶液,照“2.1”项下色谱条件下测定峰面积,以信噪比法计算,结果葛根素的最低定量限(S/N=10)为0.45μg·mL-1,检出限为(S/N=3)45.0 ng ·mL-1;大豆苷的最低定量限(S/N=10)为0.46 μg·mL-1,检出限(S/N=3)为46.0 ng·mL-1。
2.6 精密度试验 精密量取“2.3”项下的对照品溶液10μL,在上述色谱条件下重复进样6次,测定其峰面积,葛根素与大豆苷RSD分别为0.7%与0.3%,表明进样精密度良好。
2.7 稳定性试验 取供试品溶液,室温下放置,分别在0、3、6、9、12、24 h进行测定,记录葛根素与大豆苷的峰面积,计算RSD分别为0.8%和0.7%,表明葛根素和大豆苷在24 h内稳定。
2.8 重复性试验 取批号为20070112的样品,照“2.2”项下的方法各配制6份供试品溶液,按照含量测定方法平行操作,测定,结果样品中葛根素和大豆苷含量的RSD分别为0.7%与0.5%,表明本方法重复性良好。
2.9 加样回收率试验 精密量取已知含量同一批次的样品(每片含葛根素26.27 mg,含大豆苷29.18 mg),分别精密加入已知值的80%、100%、120%的葛根素与大豆苷对照品,按“2.2”项下供试品溶液的处理方法,进样记录色谱图,计算测得量,结果见表1。

表1 葛藤分散片中葛根素与大豆苷加样回收率(n=9)
2.10 系统耐用性试验 取供试品溶液,在保持其他色谱条件不变的情况下,分别改变流速(0.9、1.1 mL·min-1)、柱温(30、40℃)以及更换同类型色谱柱Xbridge C18色谱柱(4.6 mm×150 mm,5μm)以及Hypersil ODS-2色谱柱(4.6 mm×150 mm,5 μm)、流动相比例甲醇-水(25∶75,V/V)以及甲醇-水(15∶85,V/V)以考察测定条件有微小变动时,测定结果不受影响的程度。结果表明,在改变上述色谱条件下的情况下,葛根素的理论板数均大于4 000,葛根素与大豆苷及相邻峰的分离度都大于1.5,说明该方法有良好的耐用性。
2.11 样品含量测定 配置供试品及对照品溶液,照“2.1”项下色谱条件进样测定,按外标法以峰面积计算,测得三批次样品的含量,结果见表2。
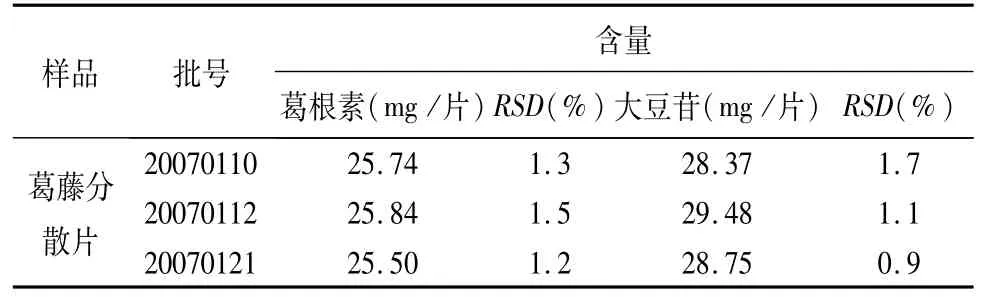
表2 三批次葛藤分散片中葛根素与大豆苷的含量测定结果(n=6)
由以上测定结果显示,本方法测定葛藤分散片中葛根素(C21H20O9)与大豆苷(C21H20O9),快速,准确,可靠。
3 讨论
3.1 检测波长的选择 在400~200 nm范围内对葛根素与大豆苷对照品进行色谱扫描,分别见图2、3,发现在葛根素在249.8 nm处有最大吸收,大豆苷在254 nm有最大吸收,因此确定测定样品检测波长为250 nm。
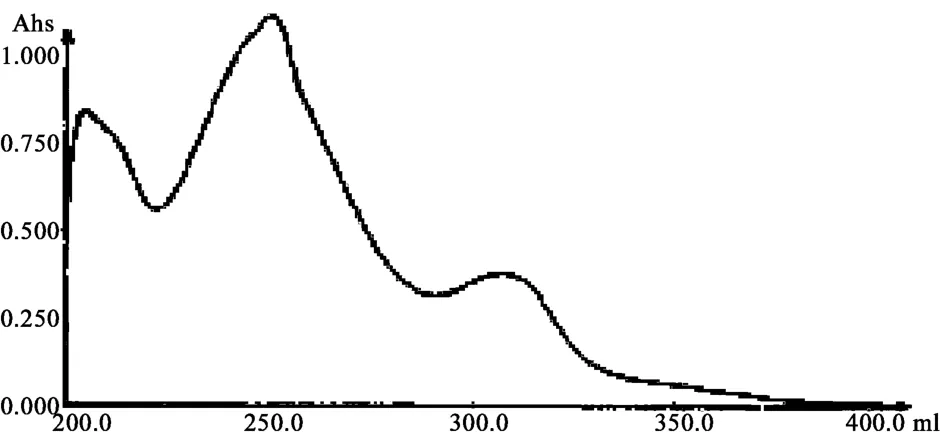
图2 葛根素对照品紫外扫描图
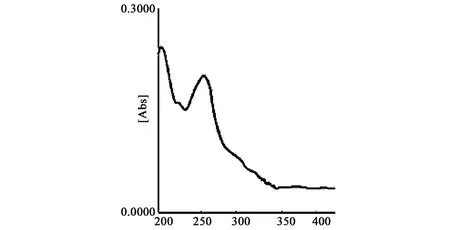
图3 大豆苷对照品紫外扫描图
3.2 葛藤分散片提取方法的考察 经过对提取方法,提取溶媒,甲醇用量及提取时间的试验验证,其葛根素与大豆苷的提取百分含量见表3。
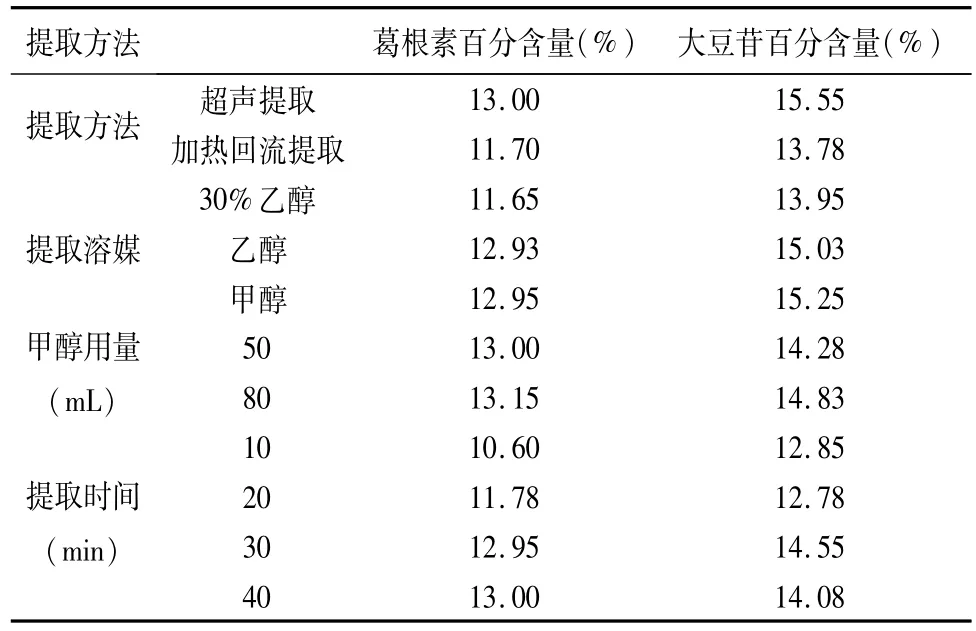
表3 葛藤分散片提取方法葛根素与大豆苷的百分含量
由表3可以确定葛藤分散片的制备方法为,取供试品(批号:20070110)10片,称量,研细,精密称取平均片重,置于250 mL具塞锥形瓶中,加入50 mL甲醇超声30 min,放冷,定容于100 mL量瓶,摇匀,精密移取过0.45μm滤膜的续滤液1 mL定容于10 mL量瓶中,摇匀,取10μL进样分析。
4 结论
该试验方法采用C18色谱柱,甲醇-水(20∶80,V/V)等度洗脱,可以快速简便的检测葛藤分散片中葛根素与大豆苷的含量,采用甲醇超声30 min,提取葛藤分散片中的葛根素与大豆苷,方法准确,便捷,提高了试验效率,试验结果表明,该方法快速、准确、可靠,可用于葛藤分散片中葛根素与大豆苷的含量测定,对其他相关中药的质量标准研究有重要的参考价值。
[1]陈荔,陈树和,刘焱文.葛根资源、化学成分和药理作用研究概况[J].时珍国医国药,2006,17(11):2305-2306.
[2]国家药典委员会.中华人民共和国药典2010年版(一部)[S].北京:中国医药科技出版社,2010:313.
[3]刘玉国,修彦凤.葛根及其葛根制剂中葛根素含量测定方法概述[J].基层中药杂志,1999,13(1):42-44.
[4]周红英,王建华,闫凤云.RP-HPLC分离测定甘葛藤茎叶中葛根素、大豆苷和大豆苷元的含量[J].中国中药杂志,2007,10(32):937-939.
[5]周红英,李同德,苏雪慧,等.野葛藤茎及叶总黄酮的含量测定[J].泰山医学院学报,2004,25(1):55.
[6]隆颖,于永洲,朱淼.HPLC测定葛根宝软胶囊中葛根素和大豆苷元的含量[J].中国医学导报,2007,4(27):101-102.
[7]周欣,宋洪涛,游开仙.梯度洗脱HPLC法测定舒肝降脂胶囊中葛根素、大豆苷及大豆苷元的含量[J].解放军药学学报,2008,24(1):78-80.
Determ ination of puerarin and daidzin in Kudzu Dispersible Tablet by RP-HPLC
DONG Hong-wei,LIUWan-hui
(School of Pharmacy,Yantai University,Yantai264005,China)
ObjectiveTo establish a RP-HPLCmethod for the determination of puerarin and daidzin in Kudzu Dispersible Tablet.MethodsAgilent Zarbax Eclipse C18column(4.6 mm×150 mm,5μm)was adopted with the mobile phase consisted ofmethanol-water(20∶80,V/V),the flow rate was 1.0 mL·min-1and the detection wavelength was set at250 nm.The column temperature was 35℃and the injection volume was 10μL.ResultsThe linear range of puerarin 9.04~72.32μg·mL-1and 9.12~72.96μg·mL-1for daidzin.The lower limitof quantification of puerarin and daidzin were 0.45 and 0.46μg·mL-1,respectively.The average recovery were 100.2%and 101.7%(n=9),respectively;RSD were 0.7%and 0.9%,respectively.ConclusionThe method was simple,specific,sensitive and accurate with good repeatability,whichmay be used for the quality control of puerarin and daidzin in Kudzu Dispersible Tablet.
Kudzu Dispersible Tablet;Puerarin;Daidzin;HPLC
R927.2
A
2095-5375(2014)02-0073-003
山东省科技发展计划项目(No.032040101)
董宏伟,男,研究方向:药物分析,E-mail:weid8848@163.com
刘万卉,男,教授,研究方向:药物分析,Tel:13306389032,E-mail:wanhui@luye.cn
猜你喜欢
润·文摘(2021年9期)2021-09-22
昆明医科大学学报(2021年4期)2021-07-23
中成药(2018年12期)2018-12-29
红岩(2018年6期)2018-11-16
中成药(2018年6期)2018-07-11
中成药(2017年10期)2017-11-16
中成药(2017年6期)2017-06-13
少儿科学周刊·儿童版(2015年1期)2015-07-07
郑州大学学报(医学版)(2015年2期)2015-02-27
青少年科技博览(中学版)(2014年4期)2014-07-31
