
红莲外皮原花青素各级分的分析鉴定
2014-02-13 01:27:20彭芳刚吴卫国李绮丽宾冬梅陈晓华
食品科学 2014年12期
彭芳刚,吴卫国,李绮丽,宾冬梅,陈晓华
(1.衡阳师范学院生命科学系,湖南 衡阳 421008;2.湖南农业大学食品科学技术学院,湖南 长沙 410128)
原花青素(proanthocyanidins,PC),是一类黄烷类单体及其聚合体的多酚类化合物,具有多种生物药理活性,如清除自由基[1]、抗氧化性[2]、防癌抗癌[3]、抗疲劳[4]等,且安全无毒性[5],在食品、药品、化妆品等领域有着广泛应用。植物体内的原花青素是一种多分散体系,聚合度从几到几十,不仅聚合度组成复杂,而且各组分间性质相近,因此除单体原花青素和部分低聚原花青素之外,很难分离纯化得到单一结构的纯多聚原花青素,只能得到一定分子质量范围的多聚原花青素混合物[6]。因此,一般只能以原花青素混合物为对象,结合化学反应和现代仪器研究化学组成和结构特征。根据红外光谱图中吸收峰谱带的位置、形状和强度,结合分子特征基团振动频率和结构的关系,可以确定分子中所含的基团或化学键,并辨别原花青素的两种单元结构——原花青定和原翠雀定[7-8]。反相高效液相色谱(reversed phase-high performance liquid chromatography,RP-HPLC)及反相高效液相色谱-电喷雾电离-电喷雾电离-质谱联用(RP-HPLC-electrospray ionization-mass spectrometry,RP-HPLC-ESI-MS),可以较好地分离原花青素单体及低聚体的某些异构体,能够确定原花青素的单体和低聚原花青素组分[9-10]。
本实验对按聚合度分级分离得到的4 种红莲外皮原花青素级分,先进行平均聚合度测定,得出各级分的平均聚合度差异,再进行红外光谱扫描,根据各级分的红外光谱图,推断红莲外皮原花青素的主要结构单元,再进行RP-HPLC分析及RP-HPLC-ESI-MS分析,揭示不同级分的组成和化学结构特征。
1 材料与方法
1.1 材料与试剂
红莲外皮粉 湖南粒粒珍湘莲食品有限公司;大孔吸附树脂AB-8 天津市海光化工有限公司;儿茶素等标准品 美国Sigma公司;乙腈和乙酸(色谱纯)、丙酮等其余试剂均为分析纯。
1.2 仪器与设备
大孔树脂层析柱(d15 mm×300 mm) 盐城泓宇玻璃仪器厂;SKY-200B恒温培养振荡器 上海沪粤明科学仪器有限公司;WFJ7200可见分光光度计 尤尼柯(上海)仪器有限公司;FD-1B冷冻干燥机 北京博医康实验仪器有限公司;Nicolent 6700傅里叶红外光谱赛默飞世尔科技公司;1290 infinity LC/6530 Q-TOF MS液-质联用系统、1100液相分析色谱仪 美国安捷伦公司。
1.3 方法
1.3.1 红莲外皮原花青素按聚合度分级分离
结合溶剂萃取法、柱层析法和溶剂沉淀法对红莲外皮原花青素按聚合度进行分级分离,分级分离流程如下。

1.3.2 红莲外皮原花青素各级分的聚合度分析
1.3.2.1 实际聚合度的测定[11]
纯原花青素的聚合度(degree of polymerization,DP)可以用相同质量的单体和聚合体产物吸光度的比值确定。

式中:A1、m1分别为单体儿茶素乙酸溶液与香草醛溶液反应后产生的吸光度和质量/g;A2、m2分别为聚合原花青素乙酸溶液反应后产生的吸光度和质量/g。
不纯的原花青素的DP计算如式(2)所示:

式中:m为原花青素质量/g;n为原花青素物质的量/mol;M为单体儿茶素的分子质量。
1.3.2.2 原花青素质量的测定
采用香草醛-盐酸法[12]。实验组:依次取0.5 mL提取液,3 mL质量分数4%香草醛-甲醇溶液以及1.5 mL浓盐酸加入试管中,充分混匀后于30 ℃水浴20 min后在波长500 nm处测定吸光度A1。对照组:依次取0.5 mL提取物溶液,3 mL甲醇以及1.5 mL浓盐酸加入试管中,充分混匀后于30 ℃水浴20 min后在500 nm波长处测定吸光度A0。空白组:蒸馏水。
原花青素的吸光度:

式中:A1平为3 次测定A1的平均值;A0平为3 次测定A0的平均值。
以儿茶素为标准品,以吸光度为纵坐标,儿茶素质量浓度(mg/mL)为横坐标,拟合得到标准曲线方程为:y=1.347 6x+0.016 6,R2=0.999 6。线性范围为0.025~1 mg/mL。依据此方程计算样液中原花青素质量浓度。
1.3.2.3 原花青素物质的量的测定[11]
取1 mL样品乙酸溶液与5 mL含4%盐酸和0.5%香草醛的乙酸溶液混合均匀,于20 ℃条件下反应5 min后在波长500 nm处测定吸光度。以乙酸代替样品溶液作空白对照。
以儿茶素为标准品,以吸光度为纵坐标,原花青素物质的量浓度(μmol/mL)为横坐标,拟合得到的标准曲线方程为:Y=5.646X+0.034 1,相关系数R2=0.999 4,线性浓度范围为0.05~0.25 μmol/mL。根据此方程计算样液中原花青素的物质的量浓度。
1.3.3 红莲外皮原花青素的红外光谱分析
将红莲外皮原花青素各级分试样与溴化钾按2%比例混合研磨压片,在红外光谱仪中先对纯溴化钾薄片进行背景扫描,然后再对含有样品的溴化钾薄片进行扫描,扫描频率为32次/s,扫描波数为4 000~400 cm-1。
1.3.4 红莲外皮原花青素的反相高效液相色谱分析及质谱联用分析
MS条件:色谱柱为C18(250 mm×4.6 mm);流动相:A为0.25%乙酸溶液,B为乙腈。梯度洗脱如下:0~20 min,2%~20% B;20~50 min,20%~35% B;50~60 min,35% B;60~65 min,35%~2% B;65~70 min,2% B。流速0.8 mL/min;柱温15 ℃;280 nm双通道紫外-可见检测器检测。
ESI-MS条件:负离子扫描方式;碎片电压135 V;毛细管电压2 500 V;雾化压力30 psi;干燥气体温度350 ℃;质谱离子范围50~1 500 D。
1.3.5 纯度计算公式

2 结果与分析
2.1 红莲外皮原花青素各级分的观察分析

图1 冷冻干燥得到的原花青素的各级分Fig.1 Freeze dried proanthocyanidin fractions
红莲外皮原花青素按聚合度分级分离后,冷冻干燥得到级分F1、F2、F3、F4,如图1所示。色泽上,F1呈淡黄色,F2、F3、F4颜色由淡红色到暗红色逐渐加深;状态上,F1一直保持粉末状态,F2、F3、F4在室温条件下放置过久会黏成一团,其中F4最容易成团,推测其原因可能是原花青素的酚羟基易与水形成羟基,所以原花青素溶液不易干燥,干燥后样品也容易吸潮[13],而且原花青素聚合度越大,越容易与水形成羟基,越容易吸潮。同时,F1、F2、F3、F4纯度经测定分别为91.86%、69.4%、55.2%、38.9%,原花青素与多糖的结合也会导致其容易吸潮。
2.2 红莲外皮原花青素各级分的聚合度分析
以原花青素级分F1、F2、F3、F4为样品,分别测定平均聚合度,结果见表1。
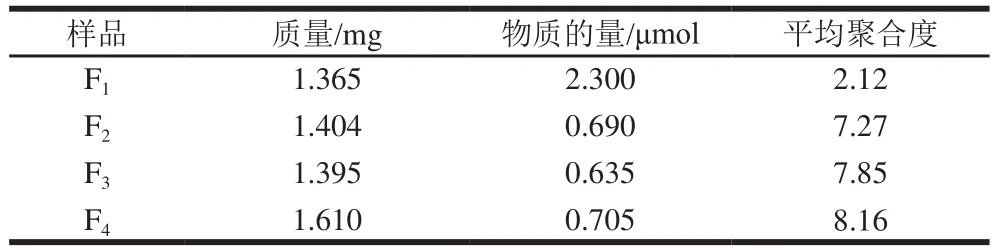
表1 原花青素各级分的聚合度Table 1 Degrees of polymerization of proanthocyanidin fractions
由表1可知,原花青素级分F1、F2、F3、F4的平均聚合度依次增大,说明通过乙酸乙酯萃取分离得到的乙酸乙酯级分(F1)为低聚原花青素,水溶性级分(F2、F3、F4)为高聚原花青素[14];采用氯仿-甲醇二相体系沉淀分离原花青素水溶性级分,依次得到级分F4、F3、F2,聚合度依次减小[15]。由此可见,采用乙酸乙酯萃取分离和氯仿-甲醇二相体系沉淀分离,使红莲外皮原花青素按聚合度大小分级分离效果明显。
2.3 红莲外皮原花青素的红外光谱分析
采用傅里叶变换红外光谱仪对F1、F2、F3、F44 种级分进行红外光谱分析,得到红外光谱图如图2所示。
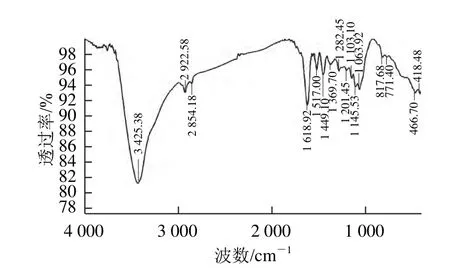
图2 原花青素级分F1的红外光谱图Fig.2 IR Spectrum of proanthocyanidin fraction F1
由图2可见,3 425 cm-1处的强吸收峰,是原花青素分子中羟基的伸缩振动,2 923 cm-1处苯环的C—H伸缩振动,1 619、1 517、1 449 cm-1处是苯环骨架C=C的特征伸缩振动峰,1 282、1 201 cm-1处是酚羟基伸缩振动及面内弯曲振动,1 146 cm-1处是C环中的C—H伸缩振动,1 103、1 064 cm-1处是原花青素分子中C—C的伸缩振动,818 cm-1处的吸收峰可能是由于苯环上有3 个相邻的H存在产生的,771 cm-1处芳香环的不饱和C—H面外变形振动。
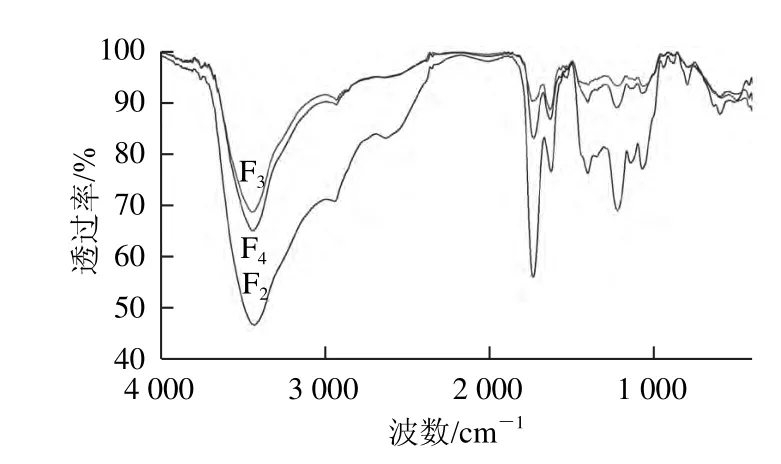
图3 原花青素级分F2、F3、F4的红外光谱图Fig.3 IR Spectra of proanthocyanidin fractions F2, F3 and F4
原花青素B环羟基的数量变化对原花青素的红外光谱有直接影响,可根据红外吸收特征粗略估计原花青定(B环含有2 个羟基)和原翠雀定(B环含3 个羟基)在多聚原花青素中的比例[16]。由图2可见,F1在强振动频率区1 540~1 510 cm-1处只有1 517 cm-1一个峰,表明F1是以原花青定为主要结构单元的原花青素或原翠雀定结构单元的比例少于60%。F1在低频指纹区780~730 cm-1处只有771 cm-1一个峰,在730 cm-1处无吸收峰,表明F1是以原花青定为主要结构单元的原花青素。由此可以推断出,F1是以原花青定为主要结构单元的原花青素。
比较图2和图3发现,F2、F3、F4的红外光谱图与F1的红外光谱图吸收峰谱带形状相似,但位置和强度不同,可能是含有的杂质使红外光谱图吸收峰偏移,即强而宽的羟基伸缩振动峰(3 400 cm-1附近),苯环的C—H伸缩振动峰(2 920 cm-1附近),苯环峰(1 630 cm-1附近),C环中的C—H伸缩振动峰(1 130 cm-1附近),原花青素分子中C—C的伸缩振动峰(1 060 cm-1附近)。F1、F2、F3、F4在2 920 cm-1和1 150 cm-1附近均有吸收峰,但F1的最尖锐,其次为F2,根据Foo[17]的研究,原花青素聚合度越大,在2 920 cm-1和1 150 cm-1附近的吸收峰越不明显,可推断F1聚合度最低,F2聚合度居中,F3、F4聚合度较高,与各级分平均聚合度的测定结果相符。
综合上述可知,F1、F2、F3、F4原花青素特征骨架振动主要集中在3 500~3 000、1 700~1 000、850~600 cm-1,且红莲外皮原花青素是以原花青定为主要结构单元。
2.4 红莲外皮原花青素的RP-HPLC-ESI-MS分析
2.4.1 原花青素级分F1的RP-HPLC-ESI-MS分析
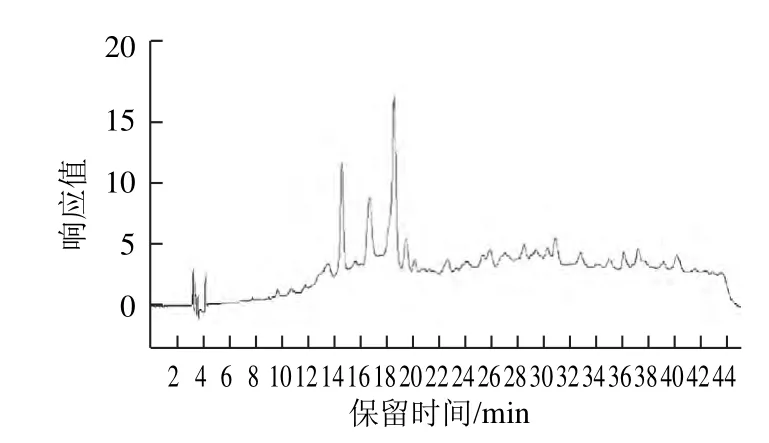
图4 原花青素级分F1的高效液相色谱图(Agilent 1290)Fig.4 HPLC chromatogram (Agilent 1290) of proanthocyanidin fraction F1
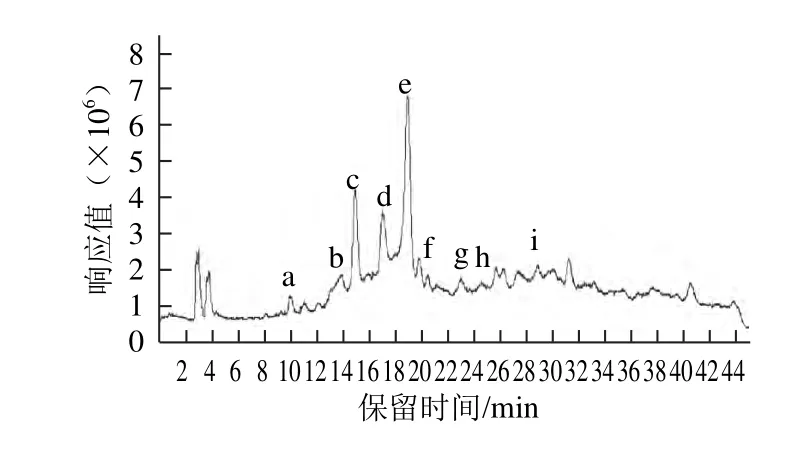
图5 原花青素级分F1的负总离子流色谱图Fig.5 Total negative ion chromatogram of proanthocyanidin fraction F1
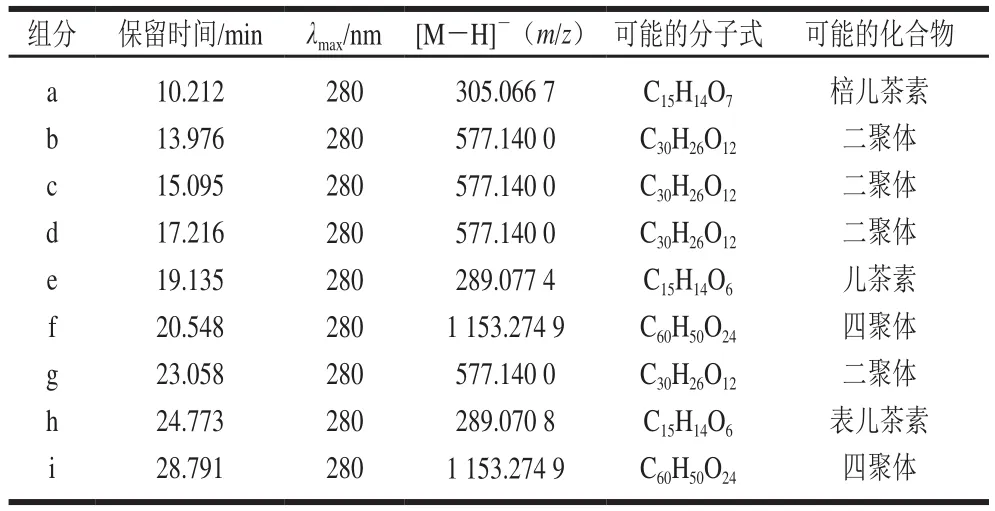
表2 原花青素级分F1的组成分析Table 2 Composition analysis of proanthocyanidin fraction F1
对原花青素级分F1进行RP-HPLC-ESI-MS分析,得到高效液相色谱图(图4)和负总离子流色谱图(图5),组成分析见表2。通过与标准品共进样,表明a(10.212 min)为棓儿茶素、e(13.976 min)为儿茶素、h(24.773 min)为表儿茶素。通过计算峰面积可知,单体中儿茶素的含量最高。推断b、c、d、g四者为由儿茶素或表儿茶素聚合而成的原花青素二聚体,构型未知;推断f、i也是由儿茶素或表儿茶素聚合而成的原花青素四聚体[18],构型未知。因此,F1的主要结构单元为由儿茶素或表儿茶素聚合的原花青定,与Ling Zhiqun等[19]对莲蓬原花青素的分析结果相同。这些数据也说明,原花青素在反相液相色谱中的出峰时间与聚合度没有必然关联[20],各单体出峰时间先后为棓表儿茶素、儿茶素、表儿茶素。
2.4.2 原花青素级分F2、F3、F4的RP-HPLC分析
采用RP-HPLC-ESI-MS分析原花青素时只能分离单体和低聚体,而更高聚合度(≥5)的原花青素,由于分子质量大,可能无法质子化,故原花青素级分F2、F3、F4不能用 ESI-MS定性分析。因此,以F1的RP-HPLC-ESIMS得出的组成分析结果为模板,来确定原花青素级分F2、F3、F4的组成。然而对F1进行RP-HPLC-ESI-MS分析时,使用的液相分析仪为Agilent 1290,而对其单独进行液相扫描时,使用的液相分析仪是Agilent 1100,两种型号的液相分析仪在同条件下扫描得出色谱图存在差异。所以使用Agilent 1100进行RP-HPLC分析时,先用单体标样确定单体的保留时间,然后根据F1的高效液相色谱图(Agilent 1290)各组分的出峰顺序及峰形,确定其他组分在F1的高效液相色谱图(Agilent 1100)中的保留时间,从而推测出这些组分是否存在于F2、F3、F4中。
标样儿茶素和表儿茶素的高效液相色谱图(Agilent 1100)见图6,其中儿茶素的保留时间为20.472 min,表儿茶素的出峰时间为23.595 min,与原花青素级分F1的RP-HPLC-ESI-MS分析得到的高效液相色谱图(Agilent 1290)和负总离子流色谱图进行比较,将上述两个峰标记为e、h,并标记组分e、h在F1的高效液相色谱图(Agilent 1100),和F2、F3、F4的高效液相色谱图(Agilent 1100)中的位置。
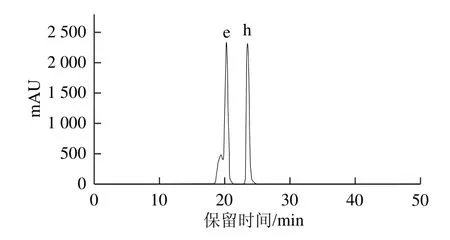
图6 标样儿茶素和表儿茶素的高效液相色谱图(Agilent 1100)Fig.6 HPLC chromatogram (Agilent 1100) of mixed standards of catechin and epicatechin
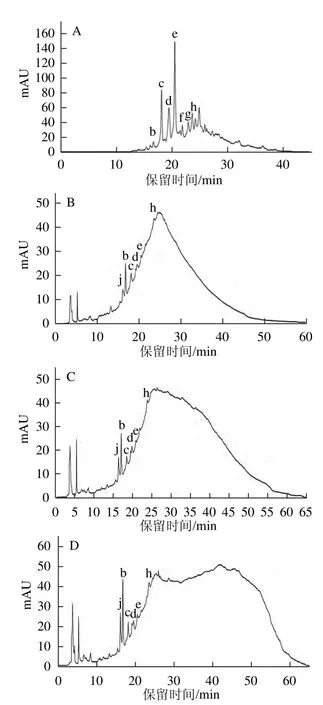
图7 原花青素级分F1(A)、F2(B)、F3(C)、F4(D)的高效液相色谱图(Agilent 1100)Fig.7 HPLC chromatograms (Agilent 1100) of proanthocyanidin fractions F1, F2, F3 and F4
通过比较F1的高效液相色谱图(Agilent 1290)和高效液相色谱图(Agilent 1100)中各组分的出峰顺序及峰形,标记组分b(16.690 min)、c(18.142 min)、d(19.504 min)、f(21.892 min)、g(23.005 min)在F1的高效液相色谱图(Agilent 1100)中的位置(四聚体i峰形无显著性特征,且距其他已鉴定的组分距离较远,无法确定位置),见图7A。再根据各组分在图7中的保留时间,标记组分b、c、d、f在F2、F3、F4的高效液相色谱图(Agilent 1100)中的对应位置,见图7B、C、D。可能由于F1中棓儿茶素的含量很低,容易损失,或受到杂质和仪器的影响,未能在上述液相色谱图中找到a(棓儿茶素)对应的峰。同时也由于F2、F3、F4含有大量高聚体,各聚合体在高效液相色谱中发生重叠,形成一个大峰而无法分离,干扰了低聚体组分的出峰,所以不能确定F2、F3、F4中是否存在组分f、g、i。但在F2、F3、F4的高效液相色谱图(Agilent 1100)中出现含量较多的新组分j(16.108 min),未能确定是何种化合物。
对图7进行整体比较分析,发现原花青素乙酸乙酯级分(F1)和原花青素水溶性级分(F2、F3、F4)峰形差别很大,已检测出的组分在F1、F2、F3、F4中的百分含量也存在很大差异。F2、F3、F4的液相色谱图在25 min前的峰形非常相似,25 min以后从F2、F3到F4峰形越来越宽,推测是含有的高聚体组分越来越多,需要更高体积分数的乙腈才能洗脱,故同梯度洗脱条件下,峰跑平时间延长。
3 结 论
采用乙酸乙酯萃取分离法和氯仿-甲醇二相体系沉淀分离法,使红莲外皮原花青素按聚合度大小分级分离依次得到F1、F2、F3、F4,其平均聚合度依次为2.12、7.27、7.85、8.16。红莲外皮原花青素的4 种级分的红外光谱图均表现出明显的原花青素特征骨架振动,且可推测出红莲外皮原花青素是以原花青定为主要结构单元的多聚原花青素。原花青素级分F1的RP-HPLC-ESI-MS分析检测出3种单体、4种二聚体和2 种四聚体,3 种单体分别为棓儿茶素、儿茶素、表儿茶素,4 种二聚体和2 种四聚体都是由儿茶素或表儿茶素单体聚合而成,其中儿茶素和2 种二聚体的含量最多。水溶性级分F2、F3、F4的高效液相色谱图与原花青素乙酸乙酯级分F1峰形差别很大,已检测出的组分在F1、F2、F3、F4中的百分含量也存在很大差异。水溶性级分F2、F3、F4均含有3 种二聚体,其中1 种二聚体含量较多,同时F2、F3、F4中还存在一种含量较多的未知组分。
[1]LUXIMON RAMMA A, BAHORUN T, SOOBRATTEE M A, et al.Antioxidant activities of phenolic, proanthocyanidin, and flavonoid components in extracts of cassia fistula[J].Journal of Agricultural and Food Chemistry, 2002, 50(18): 5024-5047.
[2]BAGCHI D, BACHI M, STOHS S J, et al.Free radicals and grape seed proanthocyanidin extract: importance in human health and disease prevention[J].Toxicology, 2000, 148(2): 187-197.
[3]ENGELBRECHT A M, MATHEYSE M, ELLIS B, et al.Proanthocyanidin from grape seeds inactivates the PI3-kinase/PKB pathway and induces apoptosis in a colon cancer cell line[J].Cancer Letters, 2007, 258(1): 144-153.
[4]邢雁霞, 刘斌钰, 邵鸿娥, 等.原花青素对小鼠耐力和抗氧化酶活性的实验研究[J].中国自然医学杂志, 2010(6): 415-416.
[5]YAMAKOSHI J, SATIO M, KATAOKA S, et al.Safety evaluation of proanthocyanidin rich extract from grape seeds[J].Food and Chemical Toxicology, 2002, 40(5): 599-607.
[6]石碧, 狄莹.植物多酚[M].北京: 科学出版社, 2000: 38-39.
[7]李春阳, 张红城, 王乃富, 等.葡萄籽原花青素的单元结构[J].江苏农业学报, 2010, 26(5): 1070-1077.
[8]WU Yanwen, SUN Qin, ZHOU Qun, et al.Fourier transform midinfrared (MIR) and near-infrared (NIR) spectroscopy for rapid quality assessment of chinese medicine preparation honghua oil[J].Journal of Pharmaceutical and Biomedical Analysis, 2008, 46(3): 498-504.
[9]肖俊松, 曹雁平, 龚玉石, 等.青蛇果原花青素分离和低聚体纯品的制备[J].食品科学, 2009, 30(17): 113-119.
[10]PRASAIN J K, PENG N, DAI Y, et al.Liquid chromatography tandem mass spectrometry identification of proanthocyanidins in rat plasma after oral administration of grape seed extract[J].Phytomedicine, 2009,16(2/3): 233-243.
[11]魏冠红, 魏作军, 苏宝根, 等.测定原花青素平均聚合度的一种新方法[J].中国食品学报, 2006, 6(6): 112-116.
[12]李绮丽, 吴卫国, 彭芳刚, 等.莲子皮原花青素测定方法的研究[J].现代食品科技, 2012, 28(2): 241-245.
[13]魏庆元.皮革鞣质化学[M].北京: 中国轻工业出版社, 1978: 376.
[14]杜晓.落叶松原花青素的分级及精细化利用研究[D].成都: 四川大学, 2006.
[15]孙芸, 孙宝才, 谷文英, 等.葡萄籽原花青素聚合度与自由基清除能力关系的研究[J].食品科学, 2007, 28(12): 423-428.
[16]孙达旺.植物单宁化学[M].北京: 中国林业出版社, 1992: 201-202.
[17]FOO L Y.Proanthocyanidins: gross chemical structures by infrared spectra[J].Phytochemisty, 1981, 20(6): 2278-2286.
[18]FRIEDRICH W, EBERHARDT A, GALENSA R.Investigation of proanthocyanidins by hplc with electrospray ionization mass spectrometry[J].European Food Research and Technology, 2000,211(1): 56-64.
[19]LING Zhiqun, XIE Bijun, YANG Erling.Isolation, characterization,and determination of antioxidative activity of oligomeric procyanidins from the seedpod of nelumbo nuciferagaertn[J].Journal of Agricultural and Food Chemistry, 2005, 53(7): 2441-2445.
[20]任其龙, 魏冠红, 金米聪, 等.反相高效液相色谱-电喷雾质谱法鉴定葡萄籽低聚原花青素[J].食品与发酵工业, 2006, 32(3): 79-82.
猜你喜欢
锦绣·上旬刊(2022年2期)2022-05-16 04:26:21
食品与发酵工业(2021年14期)2021-08-02 12:47:08
黄河黄土黄种人(2019年7期)2019-12-20 03:06:34
学与玩(2019年7期)2019-10-28 07:11:52
小雪花·成长指南(2017年6期)2017-07-03 18:16:59
创新作文(1-2年级)(2016年7期)2016-05-14 12:45:17
安徽化工(2016年5期)2016-02-27 08:25:04
天然产物研究与开发(2014年1期)2014-04-27 14:15:13
饮食科学(2014年3期)2014-03-10 09:44:02
海峡姐妹(2014年5期)2014-02-27 15:09:49
