
液相色谱–串联质谱内标法测定乳及乳制品中的三聚氰胺
2013-12-29 07:51:40刘锦丁轶聪
化学分析计量 2013年4期
刘锦,丁轶聪
(阜阳市产品质量监督检验所,安徽阜阳 236000)
三聚氰胺(melamine)是一种重要的氮杂环有机化工原料,学名三氨三嗪,广泛用于塑料、造纸、纺织、皮革、电器、涂料、医药等行业[1]。由于三聚氰胺(含氮量66%)与蛋白质(平均含氮量16%)相比含有更高比例的氮原子,所以被造假者利用添加在食品中造成蛋白质含量较高的假象[2]。我国在2008年发布并实施了《原料乳与乳制品中三聚氰胺检测方法》国家标准,其中包括液相色谱法、液相色谱串联质谱法、气相色谱串联质谱法等3 种检测方法[3],其中液相色谱串联质谱法是采用基质匹配的方法建立标准曲线,而基质匹配方法存在以下不足:(1)不同品牌奶粉基质各不相同,使用单一品种基质容易造成差异并影响检测结果;(2)因为实验人员操作稳定性问题,容易在样品处理环节引入误差,导致结果不准确。
笔者建立了一种同位素内标测定方法,既可以运用同位素内标物减少不同样品基质对检测造成的干扰,又可以避免由于人员操作造成的误差。运用该方法参加并通过了2012 年度国家认监委组织的“乳粉中三聚氰胺含量测定”能力验证活动,结果表明该方法可灵敏、准确地对乳及乳制品中三聚氰胺进行检测。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Agilent 1260 型,配备Agilent在线脱气机、自动进样器、柱温箱,美国安捷伦仪器公司;
质谱仪:Agilent 6430 型,配备电喷雾离子源,三重四级杆质谱仪,美国安捷伦仪器公司;
氮气发生器:N2LCMS–1 CLAIND 型,意大利Claind 公司;
固相萃取柱:Agilent Polymer SCX Box 型,150 mg,6 mL,美国安捷伦仪器公司;
氮吹仪:N–EVAP Ⅲ型,美国Organomation 公司;
高速离心机、超声提取仪、涡旋振荡器、微量移液器;
高纯氮气:纯度不小于99.999%;
甲醇、乙腈、乙酸铵、氨水:色谱纯;
三聚氰胺标准物质:纯度不小于99.0%,德国Dr.公司;
13C3–三聚氰胺(13C3–MEL)内标物质:纯度不小于99.0%,德国Dr.公司;
实验用水:屈臣氏蒸馏水。
1.2 液相色谱条件
色谱柱:Agilent Rx-Sil 色谱柱(50 mm×2.1 mm,1.8 μm);流动相:乙腈–水(体积比95∶5,含5 mmol/L 乙酸铵溶液),流速为0.3 mL/min;柱温:35℃;进样量:10 μL。
1.3 质谱条件
电喷雾离子源:ESI;模式:正离子;监测:多反应监测(MRM);正电压:300 V;雾化气压力:275.8 kPa(40 psi);干燥气体流速:10 L/min;干燥器温度:350℃;毛细管电压:4 000 V。
甜蜜素的定性定量离子对、碎裂电压、碰撞气能量等参数见表1。

表1 三聚氰胺监测离子及相应的碰撞能量
1.4 样品处理
称取(1.00±0.01) g 样品于50 mL 具塞塑料离心管中,加入200 μL 100 ng/mL 内标工作溶液及3 mL 水,涡旋混匀后加入7 mL 乙腈,涡旋混匀30 s 后超声提取20 min,再涡旋混匀后以5 000 r/min 转速离心10 min。依次采用5 mL 甲醇、5 mL 水对固相萃取柱进行活化,以1 mL/min 流速将离心管中上清液5 mL 加入萃取柱,弃去流出液,然后用5 mL 水和5 mL 甲醇淋洗小柱,负压抽干。用6 mol/L l5%氨化甲醇(甲醇与氨水体积比95∶5)洗脱目标物,收集洗脱液,在50℃水浴中吹干,用1 mL 乙腈复溶混合均匀后,经0.22μm滤膜过滤,待测。
1.5 实验方法
1.5.1 标准溶液制备
准确称取适量三聚氰胺标准物质,用50%甲醇水溶液配制成1.0 mg/mL 的标准储备液,将标准储备液再用50%甲醇水溶液稀释成10 μg/mL 的标准中间液,再将其用乙腈逐级稀释并加入适量内标,配制成三聚氰胺质量浓度为1.0,5.0,10.0,50.0,100.0 ng/mL的系列标准工作溶液(均含内标10 ng/mL)。
1.5.2 样品测定
仪器按1.2,1.3 条件稳定后,分别取处理后的样液和系列标准使用液各10 μL 进行测定,以面积积分,绘制标准曲线。三聚氰胺标准溶液的总离子流(TIC)谱图见图1。

2 结果与讨论
2.1 样品前处理条件
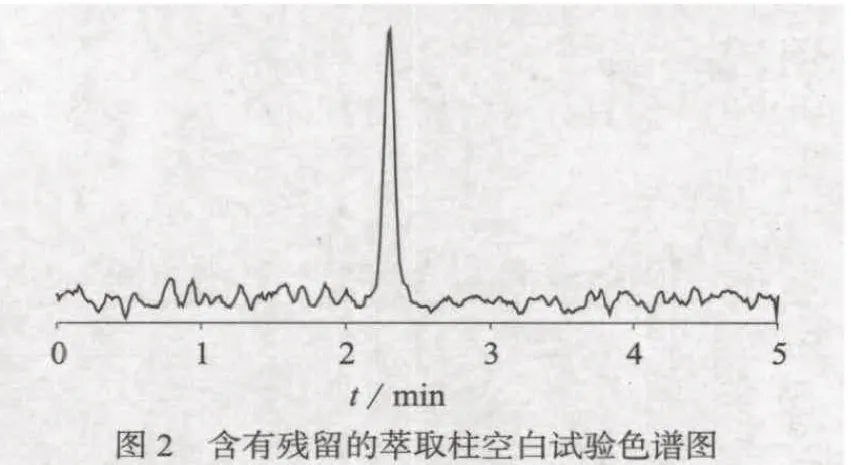
采用乙腈、甲醇水溶液、乙酸锌水溶液和亚铁氰化钾水溶液沉淀蛋白并提取三聚氰胺进行比对试验。实验证明,采用甲醇水溶液提取,回收率很低;采用乙酸锌水溶液和亚铁氰化钾水溶液沉淀蛋白时用量较大,重金属残留对色谱柱污染较严重[4]并干扰三聚氰胺的分离;乙腈沉淀效果较好,对检测基本无干扰,故选用乙腈作为提取剂。同时试验对比了Waters Oasis MCX 萃取柱和Agilent Polymer SCX Box 萃取柱,结果无明显差异,实验选择Agilent Polymer SCX Box 萃取柱。试验中发现个别萃取柱含有残留,建议实验前对小柱进行预清洗:活化后的小柱加入5%氨化甲醇淋洗,用负压抽干,置于干燥处待用。含有残留的萃取柱空白试验谱图见图2。
2.2 色谱条件的选择
根据文献[5–7]方法,三聚氰胺属于强极性的小分子碱性化合物,可以选用亲水相互作用色谱柱进行分离。实验选用的Agilent Rx-Sil 色谱柱由高纯度多孔硅胶微球制成,非常适于强亲水化合物用高有机流动相以反相模式分离。该色谱柱需要较长的平衡时间(2~3 h),否则将出现保留时间不重现的现象。在优化色谱条件过程中,分别使用了乙腈–水(体积比80∶20,含5 mmol/L 乙酸铵溶液)、乙腈–水(体积比95∶5,含5 mmol/L 乙酸铵溶液)和甲醇–水(体积比90∶10,含1 mmol/L 乙酸铵溶液)3 种体系的流动相组成,发现乙腈–水组成能够得到更好的峰形和响应值,同时分析时间最短,因此选择其作为流动相。
2.3 质谱条件的选择
根据三聚氰胺的分子结构特征,选择电喷雾离子源,在正离子模式下,首先选择全扫描模式找到适合本实验所用设备条件的三聚氰胺及其同位素内标物的母离子,然后用子离子扫描模式得到子离子,通过调整驻留时间、裂解电压和碰撞气能量得到较高的灵敏度,确定最佳实验条件。实验采用13C3–MEL作为内标物[8]进行准确定量,以消除乳及乳制品中复杂的基质干扰,保证结果的准确性。
2.4 工作曲线方程与检出限
实验表明,在1.0~100.0 ng/mL 的质量浓度范围内,以响应信号即峰面积y为纵坐标,质量浓度x为横坐标进行线性回归,得线性方程为y=1.140 255x–0.050 629,相关系数r=0.999 8。将标准品溶液稀释进样,以响应值等于3 倍基线噪声对应的三聚氰胺浓度,计算得检出限为1.0 μg/kg,比GB/T 22388–2008标准中液相色谱串联质谱法规定的0.01 mg/kg 的定量限降低了很多。
2.5 回收试验和精密度试验
采用在空白样品中添加标准溶液的方法进行回收试验,两个质量浓度添加水平分别为10,20 ng/mL,加标回收试验结果见表2。同时用添加水平为10 ng/mL 的样品重复测定6 次,测试结果 分 别 为9.980 1,9.984 4,9.992 3,9.990 4,9.978 2,9.988 9 ng/mL,计算得测定结果的相对标准偏差为0.1%。准确度和精密度高于国标要求。

表2 内标法加标回收试验结果
由表1、表2 可知,采用外标法进行加标试验,除不加内标物外,样品前处理步骤和加标量均相同,对10 ng/mL 的样品重复测定6 次,测试结果分别为7.7210,7.9561,7.0013,7.0002,7.2487,7.8864 ng/mL,相对标准偏差为5.9%,加标回收试验结果见表3。

表3 外标法加标回收试验结果
由此可见,采用内标法检测得到的测量数据的精密度和准确度均有很大程度提高,测定结果良好。
2.6 方法在能力验证中的应用
2012 年笔者采用此法参加了山西出入境检验检疫局组织的“奶粉中三聚氰胺和硫氰酸根分析能力验证”活动(编号为PT01–005),中位值为A 38.72,B 10.175,C 18.85,测试结果为A 39.84,B 10.95,C 19.41,实验室Z值的绝对值|Z|<2,结果满意。
3 结语
通过使用内标法降低了试样的基质干扰,减少了人为操作误差,对三聚氰胺的检测具有较高灵敏度,且方法的准确度和精密度较高,适用于乳及乳制品的分析检测。不足之处是同位素内标物价格昂贵,增加了检测成本,不利于方法的推广。
[1] 佘永新,柳江英,吕晓玲,等.三聚氰胺的毒性及其危害[J].食品与药品,2009,11(3): 71–72.
[2] 付登洲,杨雪娇,黄伟,等.超高效液相色谱–串联质谱内标法测定饲料中三聚氰胺含量[J].食品工业,2005(5): 59–60.
[3] GB/T 22388–2008 原料乳与乳制品中三聚氰胺检测方法[S].
[4] 李红艳,汪凤云,刘肖.离子色谱法检测含蛋白质食品中三聚氰胺[J].福建分析测试,2009,18(4): 7–8.
[5] 李新艳,王彦,谷雪,等.亲水作用毛细管整体柱的制备及其用于奶制品中三聚氰胺的加压毛细管电色谱分析[J].色谱,2010,28(3): 231–232.
[6] 梁淑明,刘冬梅,曾伟杰,等.固相萃取–亲水作用色谱法检测奶粉中的三聚氰胺[J].现代食品科技,2008,24(11): 1 180–1 181.
[7] 梅少博,侯晋,张文国,等.亲水作用色谱–串联质谱法测定化妆品中三聚氰胺残留量[J].色谱,2010,28(12): 1 189–1 197.
[8] 朱聪英,应永飞,罗成江,等.GC–MS 法同时测定生鲜乳中三聚氰胺及其类似物的研究[J].中国饲料,2011,22: 27–29.
猜你喜欢
中国化肥信息(2022年2期)2023-01-02 12:17:29
中国化肥信息(2022年8期)2022-11-30 06:20:14
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中国油脂(2020年3期)2020-04-10 02:08:54
中国化肥信息(2018年2期)2018-08-23 09:09:16
中成药(2018年6期)2018-07-11 03:01:32
无机化学学报(2016年8期)2016-12-06 09:05:14
中国化肥信息(2016年41期)2016-05-17 04:25:58
化学分析计量(2016年1期)2016-03-14 00:35:19
