
桉树组培苗潜伏期和轻基质中青枯菌的PCR快速检测
2013-12-28 04:34吴志华李国清谢耀坚周旭东
中南林业科技大学学报 2013年9期
王 艳,吴志华,李国清,谢耀坚,周旭东,2
(1.国家林业局 桉树研究开发中心,广东 湛江524022;2. 富优基尼生物技术(上海)有限公司,上海 200235)
桉树组培苗潜伏期和轻基质中青枯菌的PCR快速检测
王 艳1,吴志华1,李国清1,谢耀坚1,周旭东1,2
(1.国家林业局 桉树研究开发中心,广东 湛江524022;2. 富优基尼生物技术(上海)有限公司,上海 200235)
桉树青枯病是由Ralstonia solanacearum引起的一类土传病害,具有发病快速不易防治等特点。为了实现桉树组培苗和轻基质带菌情况的早期快速检测,将不同浓度青枯菌悬液人工接种于桉树组培苗和轻基质中,利用特异性寡核苷酸引物OLI1/Y2,能快速的检测出潜伏期组培苗,以及带菌轻基质中青枯菌的数量。试剂盒法提取基因组DNA后对其灵敏度检测结果表明:试剂盒法提取基因组DNA纯度较高,通过微量分光光度计检测出A260/A280均在1.7~1.9之间。桉树组培苗组织和轻基质中检测限分别为102CFU/mL和103CFU/mL。因此,潜伏期PCR快速检测法为预防病原菌的进一步传播提供了新的思路。
桉树青枯病;检测灵敏度;组培苗;轻基质;聚合酶链反应法
桉树青枯病是由劳尔氏菌Ralstonia solanacearum引起的能侵染50多个科200多种植物的一种细菌性土传病害[1]。1982年,在我国广西的柳桉E.saligna和巨桉E.grandis等幼树上首次报道发现[2],此后在我国广东、广西、云南、海南以及台湾等地相继报道发现。病原菌散布在土壤内, 借流水、土壤以及病残株等传播[3]。在土壤中,病原菌具有极强的存活力,即使没有寄主的存在,在适宜的温度和环境下也能长期存活[4]。病原菌可通过根的伤口或者次生根根冠部侵入寄主,然后通过木质部繁殖扩散[5],破坏植物的维管束系统,造成植物迅速失水萎蔫、枯死。同时病原菌还能定殖于植物组织内而不引起发病,与发病植株相比,潜伏期病原菌的传播存在更严重的威胁[6]。
桉树是我国重要的速生丰产林之一,自1890年引种栽培以来,我国桉树人工林面积已达360万hm2,仅次于巴西和印度,位居世界第三[7]。随着大规模桉树人工林产业的发展,高质高产的无性繁殖技术,特别是组培快繁技术越来越多的被用于优良品种的选育和规模化生产中[8]。大面积单一桉树无性系人工林的种植,导致病虫害的问题日益严重,特别是桉树青枯病的发生,给桉树生产带来了巨大的经济损失[9]。为了有效的控制病原菌的传播,建立一种桉树组培苗潜伏期及轻基质中青枯菌的快速检测方法是十分必要的。血清学技术[10]及选择性培养基[11-12]等传统的检测方法常被用于土壤及潜伏期带菌组织的检测,但是这些方法检测周期长、检测灵敏度较低及存在假阳性等缺点,不能适应植物检疫和实验室快速鉴定的需要,因此,以核酸序列分析为基础的各种分子生物学方法的建立,大大提高了对青枯菌的检测速度,这些方法可以直接从发病组织或者带菌土壤中进行病原菌的快速检测,而不需要进行病菌的分离及纯培养,同时还能在短时间内检测到极微量的病菌。简化了检测步骤的同时,也提高了检测限和准确度[13]。该研究利用Seal等[14]以16S rRNA为靶基因的青枯菌特异性引物OLI1/Y2,通过人工接种不同浓度桉树青枯菌的处理方法,从桉树组培苗及轻基质中提取出基因组DNA后并PCR检测,建立了一套桉树组培苗潜伏期及轻基质带菌灵敏度PCR快速检测方法,为防控病原菌的进一步传播提供了新的思路。
1 材料与方法
1.1 桉树青枯菌的分离、纯化及菌悬液的配制
供试菌株分离自广西省合浦县黑石岭林场尾叶桉病株,经形态学和分子生物学鉴定确定其病原菌为Ralstonia solanacearum,病原菌纯化后无菌水中室温保存。病原菌活化培养采用CPG培养基:葡萄糖5 g/L,蛋白胨10 g/L,水解蛋白1 g/L,酵母提取物1 g/L,琼脂16 g/L,调节pH值至6.5~7.0,121 ℃ 灭菌20 min待用。
无菌环境下取一环已纯化的菌株,划线于CPG培养基中,30℃培养24小时后取流动性强的菌株扩大培养36~48 h后,无菌水洗下菌体,10 000×g离心10 min,去除上清液,无菌水梯度稀释后平板计数[15]。配成10~108 CFU/mL菌悬液保存备用。
1.2 试剂与仪器
Taq DNA聚合酶;dNTPs;琼脂糖;DNeasy Plant Mini Kit购自 QIAGEN;MO-BIO PowerSoil DNA isolation Kit购自宝杰罗生物科技有限公司;Nanodorp微量紫外分光光度计。
离心机;PCR仪;电泳仪;凝胶成像系统;电泳槽;恒温培养箱等。
1.3 桉树组培苗中青枯菌DNA的提取
1.3.1 组培苗样品的处理
桉树组培苗无性系DH32-29由南方国家级林木种苗示范基地提供。取适量组培苗研磨后,分别称取50 mg研磨组织于1.5 mL离心管中,加入200 μL配制好的青枯菌悬液,使其终浓度分别为10~108CFU/mL,无菌水对照。
1.3.2 DNA的提取
试剂盒法:取处理好的不同浓度研磨组织分 别 加 入 400 μL Buffer AP1 和 4 μLRNase A,涡旋混匀后65 ˚C孵育10min,期间颠倒混匀2~3次。再加入130 μL Buffer AP2,混匀后冰上孵育5 min,20 000×g离心5 min。转移上清到QIAshredder spin中20 000×g离心2 min,转移滤液到新的离心管中,加入1.5倍体积的Buffer AP3/E混匀。转移650 μL混合液到DNeasy Mini Spin中,6 000×g离心1 min,弃去滤液,重复此步骤直至过滤完所有混合液。将此Spin放置于新的离心管中,加入500 μL Buffer AW,20 000×g离心2 min。再次转移Spin到新的离心管中,加入100 μL Buffer AE,室温孵育5 min中后6 000×g离心1 min,重复此步骤,弃去Spin,此时收集管中的DNA冷冻保存(-20℃)可直接用于下游实验。
1.4 桉树组培苗轻基质中青枯菌DNA的提取
1.4.1 轻基质样品前处理
由稻壳∶椰糠∶泥炭土等于5∶3∶2组成的桉树组培苗轻基质由由南方国家级林木种苗示范基地提供。将轻基质放于室温环境中风干后121˚C灭菌20 min待用。取灭菌基质50 mg放于1.5 mL离心管中,分别加入10~108CFU/mL的菌悬液200 μL,室温下孵育1 h。
1.4.2 DNA的提取
取处理好的样品加入到PowerBead Tubes中,轻轻涡旋混匀,加入60 μL C1溶液,上下颠倒数次混匀。将PowerBead Tubes固定在涡旋仪上,调节最大转速3 200 r/min,连续振荡10 min后室温10 000×g离心30 s。转移上清至干净的2 mL收集管中,再加入250 μLC2溶液到上清中,涡旋5 s,4℃孵育5 min后室温10 000×g离心1 min。避开沉淀小珠,转移上清到一个新的收集管中,加入200 μLC3到上清中,涡旋混匀后4℃孵育5 min。室温10 000×g离心1 min。再次转移上清到一个新的收集管中,加入1.2 mLC4溶液到上清中,涡旋混匀5 s,加载约675 μL上清到Spin Filter中,室温10 000×g离心1 min。弃去滤液后继续加载上清,重复直至过滤完所有上清。再在Spin Filter中加入500 μL C5后室温10 000×g离心30 s,弃去上清室温10 000×g离心1 min。小心转移Spin Filter到2 mL Collection Tube中,尽量避免C5溶液污染,加入100 μLC6溶液到白色滤膜中心,室温10 000×g离心30 s,弃去Spin Filter,此时收集管中的DNA冷冻保存(-20℃)可直接用于下游实验。
1.5 PCR扩增
引物设计:根据Seal等[14]结果设计引物,以16sRNA为靶基因的青枯菌特异性引物序列OLI1/Y2,由英潍捷基(上海)贸易有限公司合成。序列如下:
OLI1:5′GGGGGTAGCTTGCTACCTGCC3′
Y2 :5′CCCACTGCTGCCTCCCGTAGGAGT3′
PCR反应体系及条件:PCR采用25 μL体系:10×Buffer 2.5 μL、dNTP 0.5 μL、OLI1/Y2 各 1 μL、Taq 酶 0.5 U,模版 DNA1 μL,加 ddH2O至 25 μL。反应条件:94 ℃预变性2 min,94 ℃变性20 s,66℃复性20 s,72 ℃变性30 s,35个循环后,72 ℃延伸10 min。
琼脂糖凝胶电泳分析:1×TAE电泳缓冲液,取5 μL扩增产物于1.5%琼脂糖胶上(已加入Gel Red染色剂),60 V稳定电压分离,紫外凝胶成像分析系统观察电泳结果。
2 结果与分析
2.1 桉树组培苗中青枯菌PCR检测灵敏度的测定
将桉树组培苗研磨组织人工接种不同浓度青枯菌后,采用试剂盒法提取基因组DNA,并进行PCR扩增,电泳检测结果如图1。从图中可以看出,试剂盒法均能将菌量为102~108CFU/mL的桉树组培苗染菌组织和阳性对照扩增出单一的特异性条带,具有较高的检测灵敏度,可以检测出桉树组培苗组织中最低102CFU/mL的青枯菌。
2.2 桉树组培苗轻基质中青枯菌PCR检测灵敏度的测定
为测定桉树组培苗基质中青枯菌的PCR检测灵敏度,将不同浓度的青枯菌菌悬液人工接种于风干无菌组培苗轻基质中,试剂盒法提取基因组DNA后PCR扩增,电泳检测结果(图2)表明:PCR扩增后103~108CFU/mL处理样品及阳性对照均能扩增出单一的特异性条带,即PCR检测限最低达103CFU/mL。
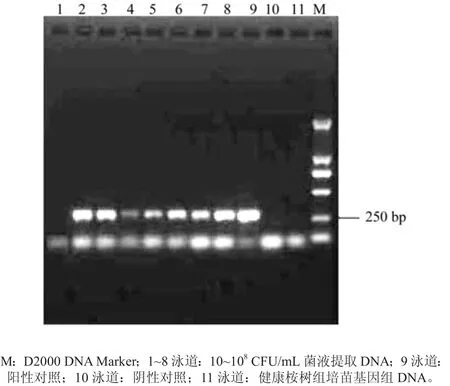
图1 桉树组培苗中青枯菌PCR检测灵敏度Fig 1 Sensitivity of PCR assay for detecting R. solanacearum in tissue culture seedlings of Eucalyptus

图2 桉树组培苗轻基质中青枯菌PCR检测灵敏度Fig 2 Sensitivity of PCR assay for detecting R. solanacearum in the substrates
3 结论与讨论
Ralstonia solanacearum是一种土壤习居菌,主要在土壤中及遗落在土壤中的病残体上越冬,也能在各种生长着的寄主体内及根际越冬,通常在土壤中存活数年之久,在湿润的土壤中可以存活二十年以上,干燥土壤中一般可以存活数十年。因此,为了进一步控制病原菌的传播,间接或直接的从自然发病植株和土壤中检测青枯菌是常用的的检测方法[16-18],但是不同的影响因素导致检测灵敏度较低[19]。随着分子生物学技术的发展,快速、灵敏且重复性较好的PCR扩增技术被广泛的用于病原菌的快速检测。其中,从植物或者土壤中提取出纯化度较高的基因组DNA是检测过程的重要步骤之一。王盛坤[20]、郝梁丞[21]等采用不同的DNA提取方法检测土壤及桉树组织中的青枯菌,均能达到较高的灵敏度,不同的DNA提取方法检测限不同[22-24]。但是综合目前的研究,由于植物组织中的酚类和多糖物质以及土壤中含量较高的腐殖酸都是影响DNA提取率和纯度的关键所在,同时对PCR的扩增产生抑制作用。因此本试验采用提取率及纯化率较高的试剂盒法,有效的提高了桉树组培苗及轻基质中青枯菌的检测灵敏度。但是此方法是否能成功的检测出自然发病组织中或自然状态下存活于轻基质中的青枯菌,还有待后续工作中的进一步研究。
利用Seal等设计的以16S RNA为靶基因的青枯菌特异性引物序列OLI1/Y2,通过人工接种青枯菌的方法,成功检测出桉树组培苗组织中102CFU/mL及轻基质中103CFU/mL的青枯菌。此方法能在12 h内完成样品的快速检测,并且具有操作简便,检测灵敏度高等优点。
为生产实践中实现大批量样品的快速检测提供了理论技术的支持。
[1] Hayward A C.Ralstonia solanacearum. In Encyclopedia of Microbiology[M].SanDiego: Academic Press, 2000:32–42.
[2] 曹季丹.巴西柳桉、巨桉青枯病调查初报[J].广西林业科技,1982, (4):30-31.
[3] Hayward A C. Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum [J]. Annual Review of Phytopathology, 1991,(29):65-87.
[4] Old K M, Wingfield M J, Yuan Z Q. A manual of diseases of eucalypts in South-East Asia [M]. Indonesia: Center for International Forestry Research, 2003:106.
[5] Xu J, Pan Z C, Prior P, et al. Genetic diversity of Ralstonia solanacearum strains from China [J]. Plant Pathology, 2009,125(4):1-13.
[6] Genin S.Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum[J].New Phytologist,2010,187(4):920-928.
[7] 杨民胜,谢耀坚,刘杰锋.中国桉树研究三十年(1981-2010)[M].北京:中国林业出版社,2011.
[8] 江海涛. 桉树组培快繁研究及其应用进展[J].现代建设,2012,11(7):64-67.
[9] Zhou X D and Wingfield M J. Eucalyptus diseases and their management in China[J].Australasian Plant Pathology, 2011,40(4):339-345.
[10] Janse J D. A detection method for Pseudomonas solanacearum in symptomless potato tubers and some data on its sensitivity and specificity[J].Bull OEPP., 1988,18(3):343-351.
[11] Granada G A, Sequeira L. A new selective medium for Pseudomonas solanacearum[J]. Plant Disease,1983,67(10): 1084-1088.
[12] Elphinstone J G, Hennessy J, Wilson J K, et al. Sensitivity of different methods for the detection of Pseudomonas solanacearum in potato tuber extracts[J].EPPO/OEPP Bulletin, 1996, 26(3): 663-678.
[13] 王 艳,常润磊,周旭东.青枯病快速检测研究概况[J].桉树科技, 2012, 29(2):53-58.
[14] Seal S E, Jackson L A, Young J P W, et al. Differentiation of Pseudomonas solanacearum, Pseudomonas syzygii, Pseudomonas pickettii and blood disease bacterium by partial 16S rRNA sequencing: construction of oligonucleotide primers for sensitive detection by polymerase chain reaction[J]. Microbiology, 1993,139(7):1587-1594.
[15] 方中达.植病研究法[M].第3版.北京:中国农业出版社,1998.
[16] González A, Plener L, Restrepo S, et al. Detection and functional characterization of a large genomic deletion resulting in decreased pathogenicity in Ralstonia solanacearum race 3 biovar 2 strains[J].Environmental Microbiology, 2011,13(12):3172-3185.
[15] Imazaki I, Nakaho K. Pyruvate-amended modified SMSA medium: improved sensitivity for detection of Ralstonia solanacearum[J]. Journal of General Plant Pathology, 2010,76(1): 52-61.
[18] Pradhanang P M, Elphinstone J G, Fox R T V. Identification of crop and weed host of Ralstonia solanacearum biovar 2 in the hills of Nepal [J]. Plant Pathology, 2002,49(4):403-413.
[19] Wilson G I. Inhibition and facilitation of nucleic acid amplification Apply Microbiology[J].Applied and environmental microbiology, 1997, 63(10):3741-3751.
[20] 王胜坤,王 军,徐大平.四种桉树青枯菌DNA提取方法及PCR检测灵敏度比较[J].中国森林病虫,2007, 26(5):4-7.
[21] 郝梁丞,赵嘉平,陶 晡,等.土壤及潜伏期桉树组织内青枯菌的PCR检测[J].河北农业大学学报,2010, 33(2):79-83.
[22] 邓 勋,宋瑞清,宋小双,等. 高效木霉菌株对樟子松枯梢病的抑菌机理[J].中南林业科技大学学报,2012,32(11):21-27.
[23] 孙志强,张兆欣,黄 琳,等. 核果类果树根癌病的药物治疗——(Ⅰ)室内效果评价[J].中南林业科技大学学报,2010,30(1): 54-58.
[24] 傅建敏,张兆欣,赵俊芳,等. 杏李杂交新品种根癌病的药物治疗II:田间效果和效益评价[J].中南林业科技大学学报,2010, 30(7):105-111.
Rapid detection of Ralstonia solanacearum in substrates and tissue culture seedlings of Eucalyptus by polymerase chain reaction method
WANG Yan1, WU Zhi-hua1, LI Guo-qing1, XIE Yao-jian1, ZHOU Xu-dong1,2
(1. China Eucalypt Research Centre , Zhanjiang 524022 , Guangdong, China; 2. Futuragene Biotechnology Co. Ltd., Shanghai 200235,China)
Bacterial wilt is rapid onset and not easy to control, and is a kind of soilborne pathogen caused by Ralstonia solanacearum.In order to realize rapid detection of the pathogens from tissue culture seedling and substrates as early as possible in latent infection,a pair of primers OLI1/Y2 was used by amplify 16S rRNA sequence to rapidly detect the pathogens and the numbers in Ralstonia solanacearum. Then, genomic DNA was extracted by using kit. The results show that the purity values of DNA A260/A280were in the 1.7~1.9; the detection limits of Eucalyptus tissue culture organization and substrates were 102CFU/mL and 103CFU/mL respectively.Therefore, the fast-detection method can provide a new way to prevent and control dissemination of bacterial wilt.
eucalyptus bacterial wilt; detection sensitivity;tissue culture seedling; light media ; polymerase chain reaction method
S792.38;S763.05
A
1673-923X(2013)09-0042-04
2012-10-16
拮抗青枯病菌菌株的筛选及应用技术(2012BAD19B08);桉树重大病虫害控制技术研究与示范(2010kjcx015-03)
王 艳(1986-),女,硕士研究生,主要研究方向:森林病害
周旭东(1973-),男,研究员,硕士生导师;E-mail:david@futuragene.com
[本文编校:吴 毅]
猜你喜欢
现代电力(2022年2期)2022-05-23
农村科学实验(2021年27期)2021-11-12
装备制造技术(2020年3期)2020-12-25
防护林科技(2020年3期)2020-05-13
儿童故事画报·发现号趣味百科(2019年9期)2019-02-02
环球时报(2019-01-03)2019-01-03
现代园艺(2018年3期)2018-02-10
制造技术与机床(2017年3期)2017-06-23
百科知识(2016年22期)2016-12-24
探测与控制学报(2015年4期)2015-12-15
