
先天性中枢性低通气综合征1例并文献复习
2013-12-26 02:00:48余永国李璧如
中国循证儿科杂志 2013年2期
任 宏 王 莹 余永国 李璧如
·论著·
先天性中枢性低通气综合征1例并文献复习
任 宏 王 莹 余永国 李璧如
目的 提高对先天性中枢性低通气综合征(CCHS)的临床和基因特征的认识。方法 总结分析1例CCHS患儿的临床表现、诊断和基因检测结果,并进行文献复习。结果 男,7月龄。以肺部感染、撤机困难入院。入院肺部感染基本控制撤机后,患儿睡眠状态下出现呼吸浅慢,再次予机械通气,模式为双水平正压通气。患儿觉醒时呼吸活跃,入睡后依赖呼吸机,自主呼吸减慢,潮气量减小,出现CO2储留。同时相关辅助检查排除了原发心、肺、脑、神经肌肉及代谢性疾病,临床诊断为CCHS。取患儿及其父母静脉血行PHOX2B基因序列检测,显示患儿PHOX2B第3外显子存在突变(基因型为20/25),其父母未检出突变,确诊为CCHS。患儿随访至11月龄,呼吸和循环情况尚平稳。结论 CCHS以觉醒时有充足通气,睡眠状态下通气不足为主要表现,行PHOX2B基因突变分析可确诊CCHS。
先天性中枢性低通气综合征;PHOX2B基因
1 病例资料
患儿,男,7月龄。14 d前出现精神疲软,嗜睡,偶咳,无明显发热,无呕吐、腹泻等表现,食欲欠佳。12 d前就诊当地医院,诊断为“呼吸道感染”,予抗感染、补液支持治疗。在输液过程出现呼吸暂停、心率下降,立即予心肺复苏,持续10~15 min后改善,收治监护病房。患儿伴有肝功能和心肌损害,肺动脉高压,经对症支持和机械通气治疗12 d后好转,期间因呼吸平稳、自主呼吸良好,予拔除气管插管,但拔管后24 h,因动脉血CO2储留明显而再次插管。患儿带气管插管转入上海儿童医学中心(我院)。
患儿系G2P1,足月剖宫产,出生体重 4 000 g ,否认产时窒息抢救史。混合喂养,按时预防接种。目前不会独坐、翻身和爬。否认食物、药物过敏史。否认手术外伤史。父母体健,否认家族性遗传病史。患儿出生23 d时曾因“新生儿肺炎”住院治疗,机械通气1月余,当时存在撤机困难。
入院查体:T 36℃,P 70·min-1,自主呼吸频率20·min-1,血压85/53 mmHg,SpO20.96(吸氧)。体重9.0 kg,头围48 cm,发育中等,面容未见特殊。胸廓无畸形,未见气促,无吸凹。两肺呼吸音粗,未闻及啰音。心律齐,心音有力,未闻及杂音。腹软,肝、脾肋下未触及。四肢水平运动良好,抬举费力,余神经系统检查未见阳性体征。
住外院期间辅助检查:血常规WBC 24.1×109·L-1,N 0.345,Hb 97g·L-1,PLT 93×109·L-1,CRP 8 mg·L-1。动脉血气分析: pH 6.9,PCO2133 mmHg,BE -9 mmol·L-1。肝、肾功能指标:ALT 1 280 U·L-1,AST 162 U·L-1,ALB 33.8 g·L-1,SCr 63.4 μmol·L-1, BUN 6.9 mmol·L-1。凝血功能检查正常。血氨101 μmol·L-1。CK 412 U·L-1,CK-MB 302 U·L-1。CSF检查:操作时有轻度损伤,色淡红,微混,RBC 10 000×106·L-1, WBC 40×106·L-1, 糖3.7mmol·L-1,氯114 mmol·L-1, 蛋白1.94 g·L-1。ECG示P波高尖,右室肥大,ST-T改变。超声心动图示肺动脉高压(72 mmHg),少量心包积液。胸、腹B超示轻度脂肪肝样改变,肝大,双侧胸腔少量积液。头颅CT示脑室少量积血,蛛网膜下隙少量出血。
入我院后辅助检查:痰、血培养阴性。血氨、电解质、血糖正常。血、尿串联质谱氨基酸谱和酰基肉碱谱正常。肌电图未见肌源性和神经源性损害。腹部B超:肝、脾和双肾未见明显异常。EEG中度异常。ECG示窦性心动过缓、不完全性右束支传导阻滞、右室大、T波变化。胸部CT示两肺感染,右上肺不张。头颅MRI示双侧侧脑室增大,脑沟加深,双侧额顶部硬膜下积液。
诊治经过:入院诊断为肺部感染,呼吸心跳骤停,心肺复苏后,心肌损害,肝功能损害。予SIMV+PSV模式机械通气,先后予头孢哌酮/舒巴坦,万古霉素,氨苄西林/舒巴坦等抗感染治疗,保肝、营养心肌等对症支持治疗,肝功能指标恢复正常,心肌损害指标改善。2次CSF检查(间隔1周)均正常。
入院1周后,患儿自主呼吸良好,血气指标正常,肺部感染控制,超声心动图未见肺动脉高压,予撤机。约36 h后,患儿睡眠状态下呼吸出现浅慢(18~20·min-1),反应差,未见三凹征等呼吸困难表现。血气分析:pH 6.838,PaCO2176 mmHg,超声心动图示肺动脉高压(59 mmHg)。再次予机械通气,以双水平正压通气模式治疗,f=0,PSV 10 cmH2O,PEEP 3 cmH2O, FiO20.25,并监测吸末二氧化碳(ECO2)变化。患儿存在觉醒时呼吸活跃,潮气量6~10 mL·kg-1;入睡后依赖呼吸机,自主呼吸呼吸减慢,潮气量减小,出现CO2储留,氧合下降(表1),需增加机械通气的呼吸频率或压力支持,才能维持正常的PaCO2。在PaCO2>60 mmHg及SpO2<0.90的情况下,未出现增加呼吸频率和(或)幅度表现。肺动脉高压好转。
鉴于患儿病情尚稳定,于入院后第24天再次拔除气管插管,在未吸氧情况下监测呼吸及氧合情况显示,觉醒时和入睡后呼吸频率分别为30~35·min-1和20·min-1,SpO2分别为>0.95和0.88~0.92。
表1 在BIPAP模式机械通气下患儿觉醒时与入睡后的呼吸参数
Tab 1 The respiratory parameters of the child during awakening and asleep under the BIPAP mechanical ventilation
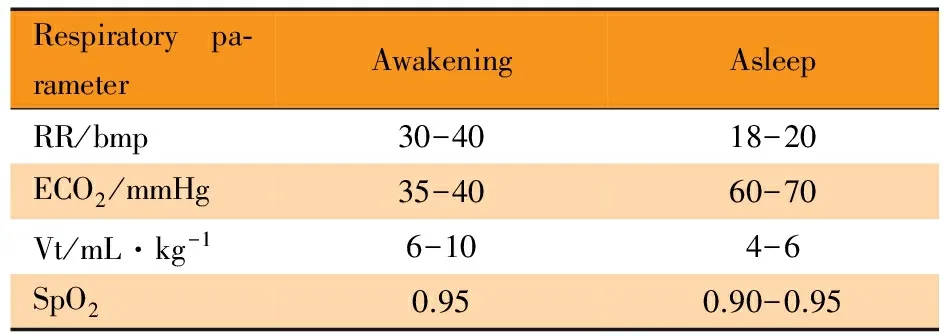
Respiratorypa⁃rameterAwakeningAsleepRR/bmp30-4018-20ECO2/mmHg35-4060-70Vt/mL·kg-16-104-6SpO20.950.90-0.95
考虑到患儿肺部感染基本控制后,仍存在撤机困难,以CO2储留为主,且入睡后出现通气功能障碍。同时相关辅助检查排除了原发心肺疾病、先天性代谢性疾病和脑干损伤,拟诊为先天性中枢性低通气综合征(Congenital central hypoventilation syndrome, CCHS)。采集患儿及其父母的静脉血,提取DNA,PCR扩增PHOX2B基因的3个外显子,之后测序。用限制性内切酶法检测100例不相关的正常对照人群以排除突变的多态性。亲子鉴定以确认家系的血源关系。结果显示,PHOX2B第3外显子存在突变(基因型为20/25),即出现25个GCN(包括GCA、GCG、GCC和GCT),比正常人多5个丙氨酸,导致蛋白质结构和功能的改变(图1),患儿父母PHOX2B基因未检测出突变。故本例确诊为CCHS。
出院后每2个月电话随访1次,患儿无呼吸道感染表现,呼吸和循环情况尚平稳,喂养正常,患儿目前随访至11月龄,会坐,不会扶站。
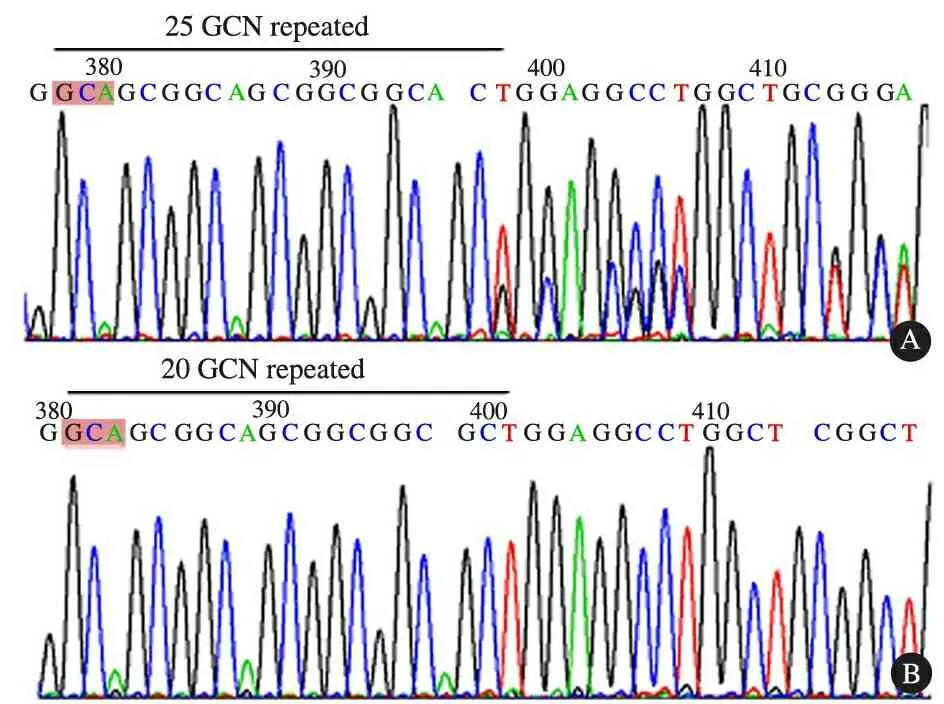
图1 本文患儿PHOX2B基因序列检测结果
Fig 1 DNA sequencing of thePHOX2Bgene in patient
Notes A: patient, B: normal control. Mutation in exon 3 of thePHOX2Bgene(genotype 20/25)
2 讨论
1970年Mellin等[1]首次将该病命名为CCHS进行了报道。截至2004年,经不完全统计,国际上相关病例报道300多例[2]。2004年广州市儿童医院报道了国内首例临床诊断CCHS患儿[3],之后陆续有10余例的临床诊断报道。根据1999年美国胸科协会的阐述,CCHS是一种最严重的呼吸和自主神经功能失调(ANSD)的表现,其中包含了呼吸控制、相关的心肺功能和自主神经功能的概念[4]。
CCHS患儿典型的临床表现以清醒时有充足通气,但睡眠状态下肺泡通气不足,出现低氧和CO2储留,而对此变化没有做出增加呼吸频率和幅度的反馈。Weese-Mayer等[2,5]分析了56例基因确诊CCHS患儿自主神经功能有关的主要临床表现,其中心血管系统如心率减慢、心律失常、迷走神经性晕厥和意识丧失的发生率为89%,呼吸系统如肺泡低通气和严重屏气发作的发生率高达100%,其他还涉及到胃肠道系统、眼部和神经系统等(表2),CCHS患儿存在一种以上的症状。Todd等[6]还对CCHS患儿的面部进行测量和比对,认为其面部特征主要表现为方短、扁平,以及可能存在手纹的改变。本文患儿入睡后出现呼吸减慢、潮气量减小、CO2储留、氧合下降,即低通气的表现,符合CCHS的典型临床表现,但未发现患儿有明显的面部特征。CCHS临床诊断的确立需首先排除原发性神经肌肉系统疾病、心肺疾病、遗传代谢性疾病及可辨认的脑干损伤。同时要排除缺氧、感染、肿瘤、外伤和梗死等合并症,本文患儿外院CSF检查由于操作时有损伤,不能以此结果来判断是否存在颅内感染;外院头颅CT提示脑室少量积血,蛛网膜下隙少量出血,可用心肺骤停、心肺复苏后来解释,之后在我院复查的头颅CT和MRI未见脑部局灶性的异常,双侧额颞部硬膜下积液是之前少量蛛网膜下隙出血的演变,符合病情的变化;且肌电图检查未有异常发现;相关检查未提示原发心肺疾病,心脏结构正常,心功能正常,肺动脉高压在改善通气后恢复正常,故符合CCHS的临床诊断。
CCHS具有明显的家族聚集和遗传的特征, 为常染色体显性遗传,PHOX2B基因是CCHS最主要的致病基因,约91%(Trang等[7])和92.6%(Trochet等[8])的CCHS患者存在PHOX2B基因突变。2003年后PHOX2B基因的检测作为确诊CCHS的手段。PHOX2B基因位于4q12,编码的蛋白含两条丙氨酸重复序列,正常情况下长的一条由20个丙氨酸重复序列组成,基因型为20/20。突变位点往往发生PHOX2B基因的第3外显子,导致丙氨酸重复序列的长度发生改变,出现24~33个丙氨酸的重复序列,即基因型为24~33/20,这是最常见一种突变情况,约占90%以上,称之为丙氨酸重复序列突变(polyalanine expansion repeat mutation,PARM),其中最常见的基因型为20/25,20/26和20/27[9,10];其他约10%的为非丙氨酸重复序列突变(non-PA repeat mutation,NPARM),包括错义、无义和移码突变。本文患儿PHOX2B基因检测结果提示基因型为20/25,为已报道的热点突变。由于PHOX2B基因编码的是一种在自主神经系统网发育中起关键作用的高度保守的同源转录因子,在人和鼠中很少发生突变,一旦出现表达的紊乱可能导致严重的功能障碍[11,12]。丙氨酸序列突变的检测对于CCHS具有诊断意义,尤其对于一些症状不典型的患儿,PHOX2B基因的检测有助于该疾病的诊断。
表2 CCHS患儿的自主神经功能相关的临床表型[2]
Tab 2 Autonomic nervous system symptoms for probands with congenital central hypoventilation syndrome and matched control subjects[2]
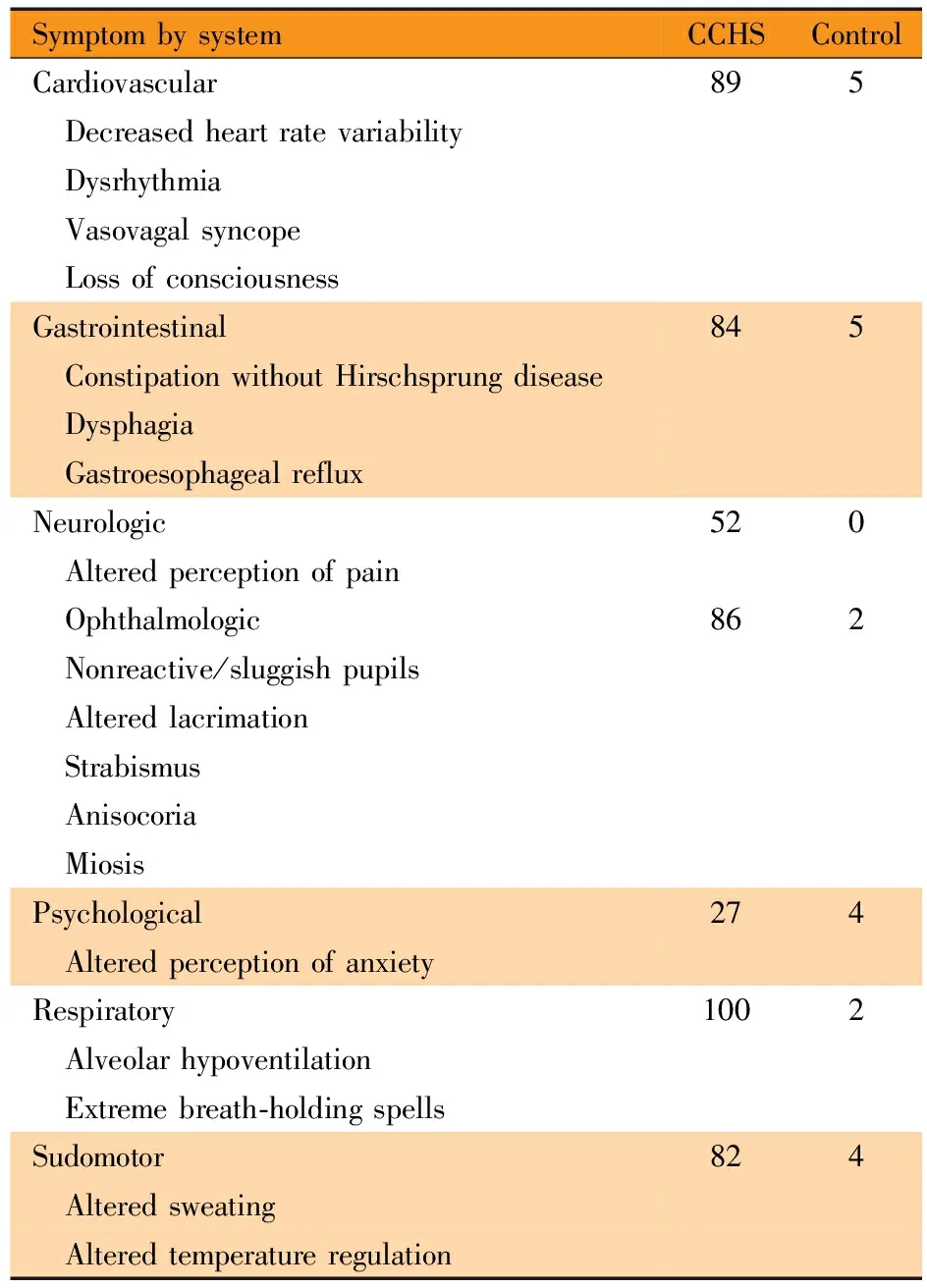
SymptombysystemCCHSControlCardiovascular895 Decreasedheartratevariability Dysrhythmia Vasovagalsyncope LossofconsciousnessGastrointestinal845 ConstipationwithoutHirschsprungdisease Dysphagia GastroesophagealrefluxNeurologic520 Alteredperceptionofpain Ophthalmologic862 Nonreactive/sluggishpupils Alteredlacrimation Strabismus Anisocoria MiosisPsychological274 AlteredperceptionofanxietyRespiratory1002 Alveolarhypoventilation Extremebreath⁃holdingspellsSudomotor824 Alteredsweating Alteredtemperatureregulation
CCHS患者PHOX2B基因型与表型之间具有一定的相关性。在PARM基因型中,约38%患者需连续通气支持,对其突变序列的长度测定,可预测患者通气功能受损的严重程度,基因型20/25和基因型20/24的成人患者,低通气的程度较轻,一般仅需在夜间进行呼吸支持。基因型20/25的患儿需要连续通气支持治疗的比例也较低,而基因型20/27~33的患儿则经常需要连续通气支持治疗。本文患儿为PARM20/25基因型,表型相对较轻,仅入睡后出现通气功能障碍,情况稳定出院后,呼吸和循环情况尚平稳。NPARM基因型CCHS患者约69%需连续通气支持,且并发巨结肠(19%vs1%)和神经嵴源性肿瘤(80%vs48%)的比例较PARM基因型高,因此通过对非丙氨酸重复序列突变的测定,可提示是否存在其他并发症[13~15]。此外,Gronli等[16]研究显示,ECG R-R间期长度与PARMs基因型的重复丙氨酸序列长度有关。同时,有研究发现PHOX2B的突变可同时存在于患儿及其父母,但仍有相当比例CCHS患儿的父母并没有携带突变的基因,提示CCHS个体存在较高的新发突变率[17]。
关于CCHS的治疗,除了对感染的控制和脏器功能的保护外,为尽可能地减少缺氧导致的损害,呼吸支持、通气治疗是关键,包括气管切开进行正压通气,无创的经鼻罩呼吸支持,膈肌起搏等[18]。由于不同的基因型对连续通气支持的依赖性不同,针对不同的患儿应给予不同的个体化治疗方案改善其预后。本文患儿入院后予连续通气24 d,病情稳定后成功脱机。Di等[19]还阐述了一系列药物在体外和体内对多聚丙氨酸延展的相关疾病,如CCHS的影响和作用。
由于CCHS的认识不足或诊断延误,间歇性的缺氧会导致年长儿神经认知缺陷、心肺功能受损和生长发育落后。因此应提高儿科医师对CCHS的认识,制定合理诊疗及随访计划,定期的评估其心、肺和脑功能,监测生长发育的情况。结合基因诊断的手段,将临床与基础研究相结合以及多学科的合作,给予CCHS患儿规范和完善的诊疗指导,可以提高生存率和生活质量。
[1]Mellins RB, Balfour HH Jr, Turino GM, et al. Failure of automatic control of ventilation (Ondine′s curse). Report of an infant born with this syndrome and review of the literature. Medicine (Baltimore),1970,49(6):487-504
[2]Weese-Mayer DE, Berry-Kravis EM. Genetics of congenital central hypoventilation syndrome: lessons from a seemingly orphan disease. Am J Respir Crit Care Med,2004,170(1):16-21
[3]Chen KZ(陈克正).Diagnosis and treatment of congenital central hypoventilation syndrome : first reported case in China.Chin J Contemp Pediatr(中国当代儿科杂志),2004,6(1):42-45
[4]Weese-Mayer DE, Shannon DC, Keens TG, et al. American Thoracic Society. Idiopathic congenital central hypoventilation syndrome: diagnosis and management. Am J Respir Crit Care Med,1999,160(1):368-373
[5]Weese-Mayer DE, Silvestri JM, Huffman AD, et al. Case/control family study of ANS dysfunction in idiopathic congenital central hypoventilation syndrome. Am J Med Genet, 2001,100(3):237-245
[6]Todd ES, Weinberg SM, Berry-kravis EM, et al. Facial Phenotype in children and young adults with PHOX2B-determined congenital central hypoventilation syndrome: quantitative pattern of dysmorphology. Pediatr Res,2006,59(1):39-45
[7]Trang H, Dehan M, Beaufils F, et al. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest,2005,127(1):72-79
[8]Trochet D, O′Brien LM, Gozal D, er al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet,2005,76(3):421-426
[9]Weese-Mayer DE, Marazita Ml, Berry-Kravis EM. Congenital central hypoventilation syndrome. Genereviews at genetests: medical genetics information resource (database online)2008. University of Washington, Seattle, 1997-2007. Available at http://www.genetests.org
[10]Loghmanee DA, Rand CM, Zhou L, et al. Paired-like homeobox gene 2B(PHOX2B) and congenital central hypoventilation syndrome(CCHS):genotype/phenotype correlation in cohort of 347 cases. Am J Respir Crit Care Med, 2009,179:A6341
[11]Brunet JF, Pattyn A. Phox2b genes-from patterning to connectivity. Curr Opin Genet Dev, 2002,12(4):435-440
[12]Ramanantsoa N, Hirsch MR, Thoby-Brisson M. Breathing without CO(2) chemosensitivity in conditional Phox2b mutants. J Neurosci, 2011,31(36):12880-12888
[13]Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic developmentand identification of mutations in PHOX2b. Am J Med Genet A,2003,123A(3):267-278
[14]Marion TL, Bradshaw WT. Congenital central hypoventilation syndrome and the PHOX2B gene mutation. Neonatal Netw,2011,30(6):397-401
[15]Weese-Mayer DE, Rand CM, Berry-Kravis EM, et al. Congenital central hypoventilation syndrome from past to future: model for translational and transitional autonomic medicine. Pediatr Pulmonol. 2009,44(6):521-535
[16]Gronli JO, Santucci BA, Leurgans SE, et al. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol,2008,43(1):77-86
[17]Berry-Kravis EM, Zhou L, Rand CM, et al. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med, 2006,174(10): 1139-1144
[18]Healy F, Marcus CL. Congenital central hypoventilation syndrome in children. Paediatr Respir Rev,2011,12(4):253-263
[19]Di Zanni E, Ceccherini I, Bachetti T. Toward a therapeutic strategy for polyalanine expansions disorders: in vivo and in vitro models for drugs analysis. Eur J Paediatr Neurol, 2011,15(5):449-452
Congenital central hypoventilation syndrome in a Chinese infant and literature review
RENHong,WANGYing,YUYong-guo,LIBi-ru
(PediatricIntensiveCareUnit,ShanghaiChildren′sMedicalCenteraffiliatedtoShanghaiJiaoTongUniversity,SchoolofMedicine,Shanghai200127,China)
REN Hong, E-mail:rhyannon75@hotmail.com
ObjectiveCongenital central hypoventilation syndrome (CCHS) is a rare autosomal dominant disorder characterized by failure in the autonomic control of breathing. MethodsThe clinical data of this patient were collected. ThePHOX2Bgene was analyzed by DNA sequecing in patient and his parents who were known with CCHS.ResultsAccording to the clinical data, this patient typically presented hypoventilation during sleeping, without any associated primary cardiac, pulmonary, neuromuscular or brainstem lesions,or any metabolic diseases. DNA sequencing of thePHOX2Bgene showed expanded alleles containing polyalanine 25 repeats in the patient.ConclusionsAccording to the clinical and genetic diagnosis, this patient presented CCHS. DNA sequencing of thePHOX2Bgene identified a mutation in exon 3 (genotype of 20/25 ) in the patient but not in his parents.
Congenital central hypoventilation syndrome;PHOX2Bgene
上海交通大学医学院附属上海儿童医学中心 上海,200127
任宏,E-mail:rhyannon75@hotmail.com
10.3969/j.issn.1673-5501.2013.02.011
2012-12-26
2013-03-09)
丁俊杰)
猜你喜欢
中老年保健(2022年5期)2022-08-24 02:35:44
疯狂英语·读写版(2022年11期)2022-05-30 08:05:00
中老年保健(2021年3期)2021-08-22 06:50:16
中华养生保健(2020年3期)2020-11-16 00:52:28
当代陕西(2017年12期)2018-01-19 01:42:06
中外医疗(2015年11期)2016-01-04 03:58:55
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
中国糖料(2013年1期)2013-01-22 12:28:23
