
Effi cacy and safety of generic escitalopram versus Lexapro in the treatment of major depression: a multi center doubleblinded randomized controlled trial
2013-12-11 05:14:18YiminYUFuafangLIBiaoWANGKeqingLIXiufengXUJianguoSFIChenggeGAOQingrongTAN
上海精神医学 2013年2期
Yimin YU, Fuafang LI*, Biao WANG*, Keqing LI, Xiufeng XU, Jianguo SFI, Chengge GAO, Qingrong TAN
·Original article·
Effi cacy and safety of generic escitalopram versus Lexapro in the treatment of major depression: a multi center doubleblinded randomized controlled trial
Yimin YU1, Fuafang LI1*, Biao WANG1*, Keqing LI2, Xiufeng XU3, Jianguo SFI4, Chengge GAO5, Qingrong TAN6
1. Introduction
Depression is characterized by high prevalence, frequent relapse, substantial disability, and increased mortality. In both high-income and low- and middle-income countries it is one of the two most important causes of disease burden.[1]The current combined prevalence of major depression and dysthymic disorder among adults in China is 4% --representing more than 35 million individuals - but only about 8% of these individuals have ever received any type of treatment for their condition.[2]One of several reasonsfor the low treatment rates is the relati vely high cost of imported proprietary anti depressant medicati ons, so the development of generic forms of anti depressants is an important step in increasing treatment rates for depressive conditi ons and, thus, reducing the huge health burden these conditi ons place on the country.
Selecti ve Serotonin Reuptake Inhibitors (SSRIs) are one important category of anti depressants. Escitalopram is an SSRI anti depressant (the S-stereoisomer of citalopram) that has been shown to have good treatment effects with relatively few side effects.[3-5]The chemical structure and treatment mechanisms of generic escitalopram produced by Jiangsu Nhwa Pharmaceuti cal Corporati on Limited are the same as those of the proprietary form of escitalopram (Lexapro) which is imported and supplied by Xi’an Janssen Pharmaceuti ca.The average monthly cost of treatment with the generic form of escitalpram is 223 Renminbi (36 US dollars)while that of the proprietary form is 501 Renminbi (81 US dollars). This study is a randomized controlled trial that aims to compare the clinical effi cacy and safety of these two forms of escitalopram.
2. Methods
2.1 Sample
The Shanghai Nental Fealth Center served as the coordinating center for the study and five other psychiatric hospitals from diff erent parts of China participated in the study. Inclusion criteria included: (a) outpati ent psychiatric pati ent at the parti cipating centers with a diagnosis of major depressive disorder based on criteria specif i ed in the Diagnostic and Stati stical Nanual of Nental Disorders IV (DSN-IV)[6](as determined by the treating clinician); (b) between 18 and 65 years of age;(c) not currently taking psychoactive medications other than sleeping medications (those previously taking medications had to be drug-free for at least 7 ti mes the half-life of the medication); (d) Familton Depression Rating Scale (FAND-17)[7]score >20 both at the ti me of screening and at the ti me of entry into the treatment phase of the study; (e) a score on the fi rst FAND item(about depressed aff ect) of >2; and (f) a score on the severity subscale of the Clinical Global Impression scale(CGI-S)[8]of > 4.
Individuals with any of the following conditions were excluded: (a) serious suicidal ideation based on the clinician’s evaluation; (b) any serious physical illness; (c)any history of epilepsy; (d) history of closed-angle glaucoma; (e) abuse of or dependence on alcohol or any psychoacti ve drug during the past year; (f) depressive episode induced by other mental or physical illnesses;(g) lactation, current pregnancy, or any possible pregnancy during the trial; (h) history of severe drug allergy;or (i) a history of poor response to escitalopram.
The enrollment of subjects is shown in Figure 1. A total of 260 individuals were recruited from Narch 13 to October 7, 2009 and 130 individuals were randomly assigned to the study group (those using generic escitalopram) or the control group (those using Lexapro).The study group included 46 males and 84 females; their mean (s.d.) age was 37.2 (12.9) years; 37 (28.5%) had a college education; 53 (40.8%) were in their first episode of illness, 58 (44.6%) had had multiple depressive episodes, and 19 (14.6%) had chronic depression; the median (intraquarti le range) duration of the current episode of depression and the total duration of depressive episodes were 3 (1 to 4) months and 15 (4 to 48) months, respectively; and 80 (61.5%) reported that‘psychological pressure’ was the main precipitant of their current depressive symptoms. The control group included 49 males and 81 females; their mean age was 39.4 (12.8) years; 43 (33.1%) had a college education;59 (45.4%) were in their first episode of illness, 51(39.2%) had had multiple depressive episodes, and 20(15.4%) had chronic depression; the median duration of the current episode of depression and the total duration of depressive episodes were 3 (1 to 6) months and 16 (6 to 55) months, respectively; and 69 (53.1%)reported that psychological pressure was the main precipitant of their current depressive symptoms. No statistically significant differences were found between the groups for sex, age, level of education, duration of current episode, total duration of depressive episodes,type of depressive episode, or reported triggers of the depressive episode. Before treatment, the mean scores on the FAND-17 were 24.7 (3.2) and 24.7 (3.3) for the study group and control group, respectively (t=0.04;p=0.970).
2.2 Study design
This is a multi center double-blind randomized controlled trial comparing the efficacy and safety of generic escitalopram and its commercial counterpart, Lexapro,in the treatment of major depressive disorder. At each of the six centers two or three research clinicians were trained in the protocol and in the administration of the evaluative instruments employed in the study. Standard methods were employed to ensure maintenance of blinding; only the study coordinator at each site (who was not involved in the evaluation or treatment of subjects) was able to break the blind during the course of the study. Strati fi ed randomization (stratified by institution) was used to assign parti cipants to the studyor the control group using the ‘Drug and Stati sti cs’(DAS) soft ware version 2.1.1 (a Chinese stati sti cal package). The total duration of observation was eight weeks. Both generic escitalopram (10 mg/tablet, batch number, 20071001) and Lexapro (10 mg/tablet, batch number, 2163474) were taken orally once a day. (These medications were all provided by the Jiangsu Nhwa Pharmaceuti cal Corporati on Limited.) The initi al dosage for both groups was 10 mg/d. At the end of the second week the dosage was increased to 20 mg/d if the pati ent tolerated the medicati on well but the treatment eff ect was considered poor (i.e., a score of >3 on the improvement subscale of the Clinical Global Impression scale [CGI-I][8]) or if the treati ng clinician thought it necessary to increase the dosage. Alternatively, the dosage was maintained at 10 mg/d aft er two weeks of treatment if the treatment was considered effective(e.g., CGI-I <3), if the treating clinician did not think it necessary to increase the dosage, or if the patient did not tolerate the medicati on well.

Figure 1. Flowchart of the study
During the trial, sleep medications (e.g., zolpidem,zopiclone, midazolam, alprazolam, clonazepam, and estazolam) at usual dosages were allowed before the patient went to sleep. The maximum durati on of consecuti ve treatment with sleep medicati on was one week. No other forms of treatment that could potentially interfere with the treatment outcome were allowed, including anti psychotics, anti depressants,anti -anxiety medications, mood stabilizers, systematic psychological therapy, or electroconvulsive therapy.
Pati ents were withdrawn from the study if any of the following occurred: (a) patient or family request to withdrawal (usually due to poor treatment effect or severe side effect); (b) discontinuation of medication for at least three consecutive days; (c) loss to followup; (d) treating clinician recommends withdrawal from study (usually due to poor compliance or the occurrence adverse events); (e) break of blinding(i.e., treating clinician or patient knows which type of medication is being administered); (f) the occurrence of manic or psychotic symptoms for at least two weeks; (g)the occurrence of a suicide attempt; (h) poor treatment effect or exacerbated symptoms after four weeks of treatment; or (i) pregnancy during the trial.
This study was approved by the insti tuti onal review board of the Shanghai Nental Fealth Center.
2.3 Evaluati on of treatment effect and safety
The main index for treatment eff ect was the change in the FAND total score at the end of eight weeks of treatment. Clinical remission was considered when the total FAND score was equal to or less than seven.Treatment was considered eff ecti ve when there was a 50% or greater reduction in the baseline FAND total score; treatment was considered ineffecti ve if the FAND reducti on was less than 50%. Secondary outcome measures included the Nontgomery-Asberg Depression Scale (NADRS),[9]the Familton Anxiety Scale (FANA),[10]and changes in the CGI score. The validity and reliability of Chinese versions of FAND,NADRS, and FANA are sati sfactory.[11]These outcomes were assessed by the treati ng clinician at baseline, and at the end of the 1st, 2nd, 4th, and 8thweeks of treatment(or at the ti me of termination from the study).
Evaluati ons of safety included assessment of vital signs and identification of adverse events by asking patients about the occurrence of any physical or psychological changes (whether or not they are related to medication use) at the end of the 1st, 2nd, 4th, 6thand 8thweeks of treatment. Laboratory tests of blood and urine and electrocardiograms were conducted at baseline and at the end of the 8thweek of treatment.
2.4 Stati sti cal analysis
The required sample sizes were esti mated using standard methods for tests of non-inferiority (equivalence trial). Based on data reported from the company that developed escitalopram, the total FAND score dropped 12.3 points aft er eight weeks of treatment. Using 12 as the average required drop in FAND scores, a δ (non-inferiority index) of 2.5, a type I error of 0.025, a type II error of 0.2, and an overall standard deviation in the mean before versus aft er FAND change score of 6, the calculated sample size for each group was 90. Considering the requirement of the national Law about the registration of new drugs and potential loss of follow-up,the final sample size was set at 260, 130 in each group.
SAS 9.1.3 was used for all data analysis. Two-sample and one-sample t-tests were used to compare continuous variables and chi-square tests were used to compare categorical variables. Analysis of covariance was used to analyze changes in the main outcome measure (the FAND-17 total score) aft er treatment with the center and group assignment as covariates. Repeated measures analysis of variance was used to compare the change in outcome measures between the groups over the fivee valuation points (baseline and at the end of the 1st, 2nd, 4th, and 8thweeks of treatment). The Full Analysis Set (FAS) and Safety Set (SS for safety analysis)included all 260 individuals enrolled in the treatment phase of the study; that is, they were ’Intenti on-to-Treat’ (ITT) analyses in which the Last Observati on Carried Forward (LOCF) imputati on method was used. The Per Protocol Set (PPS) included 210 individuals, including 193 individuals who completed all eight weeks of treatment and 17 individuals who dropped out before completi on of eight weeks of treatment but had achieved remision criteria prior to dropping out (their final assessment results were used to impute values at subsequent evaluation periods).
3. Results
3.1 Recruitment and completion
A total of 260 individuals were enrolled in the study with 130 in either arm. There were 35 (26.9%) dropouts in the study group and 32 (24.6%) in the control group (χ2=0.18, p=0.671). Figure 1 provides a list of the reasons for dropout in both groups.
3.2 Treatment effect
3.2.1 Changes in the HAMD total score and test of the null hypothesis

?
?
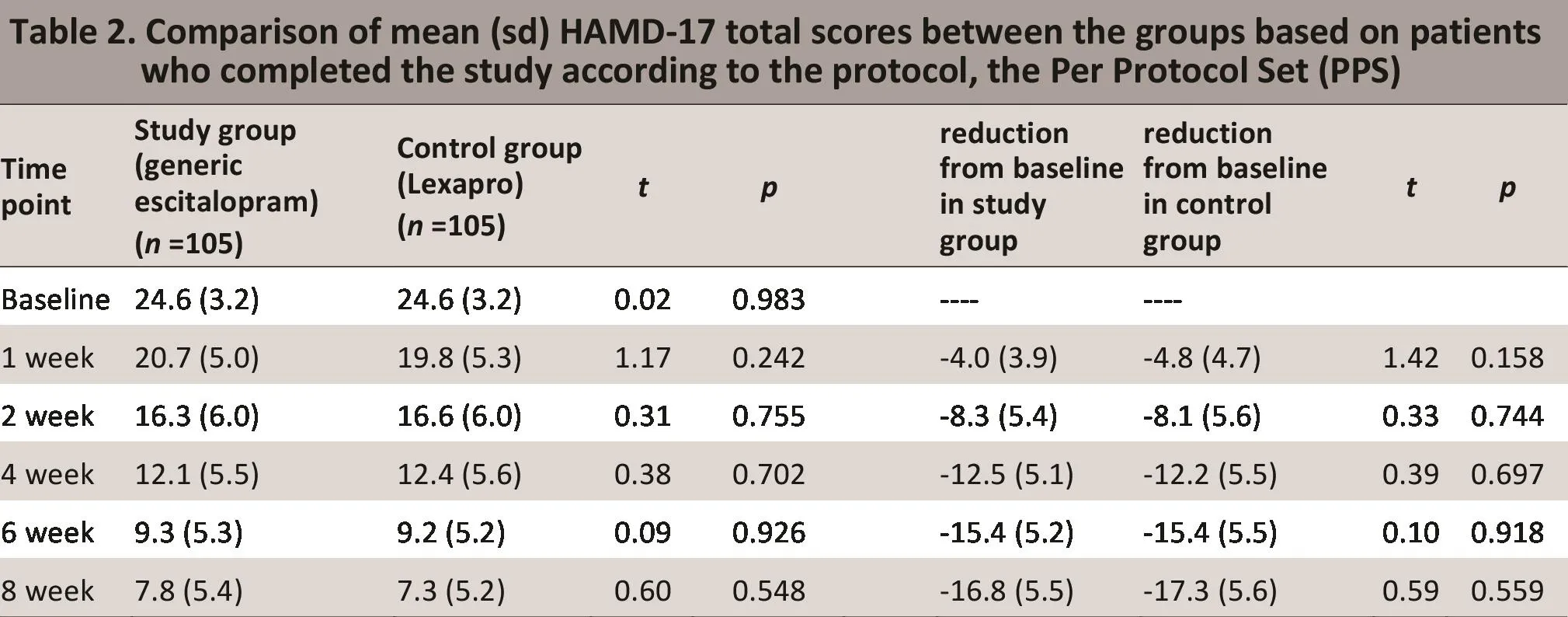
As shown in Tables 1 and 2, FAS analysis and PPS analysis found no statistically significant differences in the reduction from baseline of the FAND scores at any of the four follow-up ti me periods used in the study. The overall ti me trend analysis also showed no statistically significant difference between the two groups either in the FAS analysis (F=0.44, p=0.664) or in the PPS analysis(F=0.59, p=0.559). The mean diff erence in the FAND total scores between the two groups (study group - control group) at week 8 was 0.43 (95% confidence interval[CI]=-1.55, 2.41) for the FAS analysis and 0.43 (95% CI=-0.96, 1.81) for the PPS analysis. As shown in Table 3,aft er adjusting for baseline FAND scores and for the six treatment centers using an analysis of covariance, the difference between groups in the FAND total score at the end of 8 weeks of treatment remained statistically insignificant for both the FAS and PPS analyses. Noreover, the proportion of subjects who reached remission criteria (total FAND score <7) and the proporti on in which the treatment was considered effective (>50%reducti on from baseline FAND score) aft er 8 weeks of treatment were not signif i cantly different between the two groups: in the FAS analysis, 50.8% (66/130) and 49.2% (64/130) achieved remission in the study and control groups, respectively (χ2=0.06, df=1, p=0.804);and in 69.2% (90/130) and 66.9% (87/130) of the study and control group subjects, respectively, the treatment was considered effective (χ2=0.16, df=1, p=0.690).
3.2.2 Comparisons of MADRS, HAMA, and CGI-I scores
Analysis of the FAS showed that eight weeks after the beginning of the treatment, the NADRS total score decreased from 30.2 (6.1) at baseline to 8.0 (5.9) in the study group and from 30.9 (6.3) to 8.1 (6.8) in the control group (t=1.12, p=0.902). Similarly, the FANA score decreased from 21.0 (5.2) at baseline to 6.2 (3.9) in the study group and from 20.4 (5.8) to 6.0 (4.6) in the control group (t=0.43, p=0.669). Aft er 8 weeks of treatment the proporti ons of ‘substanti ally improved’ and ‘improved’ on the CGI-I were 64.2% and 27.4% in the study group and 66.3% and 24.5% in the control group (t=0.14, p=0.886).

?
3.3 Safety
3.3.1 Occurrence of adverse events
During the trial 59 (45.4%) individuals in the study group and 74 (56.9%) in the control group experienced one or more adverse events (χ2=3.46, p=0.063). Nost of these adverse events were mild to moderate in severity and of relatively short duration but in 9 (6.9%)individuals in the study group and 8 (6.2%) individuals in the control group these adverse events were severe enough, persistent enough or distressing enough to require withdrawal from the study (χ2=0.06, p=0.802).Four severe adverse events occurred in the study group during the trial (one case of lung cancer, two suicide attempts, and one suicide death) and one severe adverse event occurred in the control group (a suicide death) (Fisher Exact Test p=0.176). In the study group the suicide occurred on the 35thday of treatment and the two suicide attempts occurred on the 2ndday and 20thday of treatment; in the control group the suicide occurred on the 6thday of treatment. Only one of these severe adverse events (the suicide death in the study group) was considered by the treating clinician as possibly related to the use of study medication.
The adverse events with an occurrence of greater than 1% in the study group (i.e., that occurred in at least two different individuals) included dry mouth(12.3 %), nausea (9.2%), dizziness (6.2%), decreased appetite (5.4%), drowsiness (5.4%), fatigue (3.8%),insomnia (3.1%), headache (3.1%), diarrhea (2.3%),heartburn (2.3%), constipation (2.3%), excessive sweating (2.3%), irregular heartbeat (2.3%), pressure in the chest (2.3%), stomach bloating (1.5%),stomach discomfort (1.5%), upper respiratory infection (1.5%), feeling of distention in the head(1.5%), excessive yawning (1.5%), palpitation (1.5%),overall physical discomfort (1.5%), and suicide attempt (1.5%). The majority of adverse events were in the digestive system (37.7%) or the neurological system (27.7%).
In the control group adverse events with an occurrence of greater than 1% included nausea(10.8%), fatigue (7.7%), drowsiness (6.9%), dizziness(5.4%), dry mouth (4.6%), decreased appetite (4.6%),diarrhea (3.8%), constipation (3.8%), headache(3.8%), palpitation (3.8%), discomfort in the stomach(3.1%), upper respiratory infection (2.3%), fainting(2.3%), sleepiness (2.3%), urine track infection (2.3%),abdominal pain (1.5%), insomnia (1.5%), sprain in the right leg (1.5%), delayed urination (1.5%), and formication (1.5%). The majority were events in the digestive (36.2%) or the neurological system (34.6%).
3.3.2 Lab tests, vital signs and physical examinations related to safety
Clinically significant changes in blood and urine tests were identified at the end of the 8thweek of treatment in 8 (6.2%) individuals from the study group and 12 (9.2%) from the control group (χ2=0.87,p=0.351). In the study group one individual had an elevated white blood count, one had elevated neutrophils, one had urinary leucocytes, two had elevated alanine aminotransferase (ALT), and three had elevated aspartate aminotransferase (AST). In the control group two individuals had and elevated white blood cell count, one had a decreased white blood count, one had elevated neutrophils, one had a low hemoglobin, one had urinary protein, one had white blood cells in the urine, one had elevated ALT,one had elevated AST, and two had elevated fasting blood glucose. No serious physical consequences were observed among study participants who had these changes.
No statistically significant changes or abnormalities in heart rate, in the systolic and diastolic blood pressure, or in the electrocardiogram results were observed during the treatment in either of the groups.
4. Discussion
4.1 Main fi ndings
In this study, the baseline FAND-17 score was 24,indicating that the majority of patients were moderately to severely depressed. The magnitude of the mean drop in the total FAND score after 8 weeks of treatment of 13.9 (8.2) in the study group was similar to that found in other studies of escitalopram.[12]The proporti ons of subjects in whom the treatment was effective (69% in the study group and 67% in the control group) and the proportions who achieved clinical remission (51% in the study group and 49% in the control group) were also in line with the findings of other studies.[13]Our results indicate that both the generic and trade name forms of escitalopram are efficacious anti depressants and that there are no significant differences in the efficacy of the two forms of the medication.
Some studies from other countries have found that escitalopram has a good treatment effect on anxiety symptoms.[14,15]This is supported by findings from the current study. Aft er 8 weeks of treatment, the FANA score decreased significantly in both groups. The magnitude of the reduction in anxiety symptoms was not significantly different between the two forms of escitalopram.
Common adverse events in both groups were dry mouth, nausea, dizziness, drowsiness and decreased appetite; thi spat tern of adverse eff ects is similar to that reported in other studies of escitalopram.[12,16]
The occurrence of four suicidal events (two fatalities and two attempts) in the 260 enrolled subjects (1.5%,95% CI=0.4, 3.9%) during 8 weeks of treatment despite excluding patients considered at high risk of suicide from the study was concerning. Three of the four suicidal events occurred in the first month of treatment and two of them occurred in the first week of treatment. Suicidal behaviors are rare, but depressed persons are certainly at elevated risk of suicide and there is some evidence that suicidal risk is highest during the first month of anti depressant treatment, so this rela-tively high rate could be a statistical outlier.[17]Nevertheless, larger studies in which subjects are followed over longer periods are needed to determine whether the rates of suicidal events are higher in those treated with escitalopram than in those treated with other an-ti depressants. And further efforts are needed to better identify and effectively intervene with the minority of depressed patients who are at high risk of suicide.
A meta-analysis comparing effectiveness and safety of 12 commonly used anti depressants found that generic escitalopram was one of the anti depressants with the best treatment effects and one of the highest rates of treatment adherence.[18]Results from this study conf i rm this finding in Chinese patients with moderate to severe episodes of major depression who seek treatment in psychiatric outpatient services.
4.2 Limitations
The current study is a multi -center study that enrolled outpatients with depression from six psychiatric hospitals in China. It is unclear how many patients were screened at the centers to identify the 286 potential participants so it is not possible to be certain of the extent to which recruited patients were representative of all depressed individuals in China who receive psychiatric treatment. Noreover, the results could be different in a sample of patients treated at nonspecialty hospitals, who typically have less severe forms of depression than those treated at psychiatric hospitals. The sample size, though sufficiently large to assess the main hypothesis about the similarity of the two forms of the medication, was not large enough to conduct stratified analysis (e.g., by gender, age group,number of previous depressive episodes, etc.) so there may be some differences in the outcomes of the two forms of the medication in subgroups of subjects that were not identified. The follow-up period was only 8 weeks, so it is not possible to say anything about the comparability of the two forms of medications during prolonged usage or during the maintenance phase of the treatment of depression. The dropout rate of 27%in the study group and 25% in the control group was relati vely high but not out of line with what is seen in similar studies. Additional work is needed to identify biological markers that can identi fy which pati ents will respond best to which anti depressants.
4.3 Significance
This study compared the efficacy and safety of generic and brand name escitalopram. In summary, the efficacy and safety of the generic form of escitalopram (produced by Jiangsu Nhwa Pharmaceutical Corporation Limited)during the fi rst 8 weeks of treatment of a depressive episode are not significantly different from the efficacy and safety of the brand name form of escitalopram(Lexapro, produced by Xi’an Janssen Pharmaceuti ca)when used at a dosage of 10 to 20 mg per day in moderate to severely depressed pati ents treated in the outpatient departments of psychiatric hospitals in China. Both forms of the medicati on are effective and safe and they are also both relatively effective in alleviating the anxiety symptoms that commonly co-occur with depression. The occurrence of 4 suicidal events (2 deaths and 2 attempts) in the 260 patients(1.5%) treated with escitalopram during the first 35 days of treatment may be a statistical outlier, but the rates of suicidal events should be re-assessed in larger studies with longer follow-up ti mes.
Conf l ict of interest
The authors report no conf l ict of interest related to this manuscript.
Funding
Funding for this study was received from three sources:(a) the Nati onal Science and Technology Najor Project for Investi gati onal New Drugs subproject titled ‘Clinical Technological Plaる orm for Evaluati on of New Drugs in Psychiatry’ (CPEP number 2012ZX09303-003); (b) the Shanghai Jiao Tong University School of Nedicine 985 Project ti tled ‘Standardized Plaる orm for Clinical Testing of Neuropsychiatric Nedicati ons’; and (c) the Jiangsu Nhwa Pharmaceuti cal Corporati on Limited.
1. Nurray CJL, Vos T, Lozano R, Naghavi N, Abraham D.Flaxman AD, et. al Global Burden of Disease Study Diseases and Injuries Group. Disability-adjusted life years (DALYs)for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380(9859): 2197-2223.
2. Phillips NR, Zhang JX, Shi QC, Song ZQ, Ding ZJ, Pang ST et. al. Prevalence, associated disability and treatment of mental disorders in four provinces in China, 2001-2005: an epidemiological survey. Lancet 2009, 373:2041-2053.
3. Feighner JP, Overo K. Nulti center, placebo-controlled, fixeddose study of citalopram in moderate-to-severe depression.J Clin Psychiatry 1999; 60(12):824-830.
4. Wagner KD, Robb AS, Findling RL, Jin J, Guti errez NN,Feydorn WE. A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescent. Am J Psychiatry 2004; 161(6):1079-1083.
5. Kasckow JW, Welge J, Carroll BT, Thalassinos A, Nohamed S.Citalopram treatment of minor depression in elderly men:an open pilot study. Am J Geriatr Psychiatry 2002; 10(3):344-347.
6. American Psychiatric Association. DSM IV Sourcebook.Virginia: Am Psychiatric Press Inc; 1994.
7. Familton N. Development of a rati ng scale for primary depressive illness. Br J Soc Clin Psychol 1967; 6(4): 278-296.
8. Guy W. ECDEU Assessment Manual for Psychopathology.Rockville, ND: U.S Nati onal Insti tute of Fealth,Psychopharmacology Research Branch; 1976: 534-537.
9. Nontgomery SA, Asberg N. A new depression scale designed to be sensiti ve to change. Brit J Psychiatry 1979;134 (4): 382-389.
10. Familton N. The assessment of anxiety states by rating. Br J Med Psychol 1959; 32(1): 50-55.
11. Zhang NY. Handbook of Rating Scales in Psychiatry. 2nded.Changsha: Funan Science and Technology Press; 1998. (in Chinese)
12. Ou JJ, Xun GL, Wu RR, Li LF, Fang NS, Zhang FG. Efficacy and safety of escitalopram versus citalopram in major depressive disorder: a 6-week, multi center, randomized,double-blind, fl exible-dose study. Psychopharmacology(Berl)2011; 213(2-3): 639-646.
13. Schmitt L, Tonnoir B, Arbus C. Safety and effi cacy of oral escitalopram as conti nuati on treatment of intravenous citalopram in pati ents with major depressive disorder.Neuropsychobiology 2006; 54(4): 201-207.
14. Davidson JR, Bose A, Wang Q. Safety and efficacy of escitalopram in the long-term treatment of generalized anxiety disorder. J Clin Psychiatry 2005; 66(11): 1441-1446.
15. Waugh J, Goa KL. Escitalopram: a review of its use in the management of major depressive and anxiety disorders.CNS Drugs 2003; 17(5): 343-362.
16. Chen FY, Li FF, Kuang WF, Zhao JP, Xie SP, Tao N.Randomized double-blind controlled trial of escitalopram versus citalopram in the treatment of depression. Shanghai Archives of Psychiatry 2010; 22(5): 301-303. (in Chinese)
17. Jick F, Kaye JA, Jick SS. Anti depressants and the Risk of Suicidal Behaviors. JAMA 2004; 292(3): 338-43.
18. Cipriani A, Furukawa TA, Salanti G, Geddes JR, Figgins JP,Churchill R. Comparative efficacy and acceptability of 12 new-generati on anti depressants: a multi ple-treatments meta-analysis. Lancet 2009; 373(9665): 746-758.
国产草酸依地普仑片与来士普治疗抑郁症有效性和安全性:随机双盲、对照、多中心临床研究
余一旻1李华芳1* 王飚1* 栗克清2许秀峰3师建国4高成阁5谭庆荣6
1上海交通大学医学院附属精神卫生中心 上海
2河北省精神卫生中心 河北保定
3昆明医学院第一附属医院 云南昆明
4西安市精神卫生中心 陕西西安
5西安交通大学附属第一医院 陕西西安
6第四军医大学西京医院 陕西西安
李华芳: lhlh5@yahoo.com.cn; 王飚: wangbaio@yahoo.com.cn
背景抑郁症已日益成为影响国人健康的公共卫生问题,但只有少部分抑郁症患者获得治疗。治疗率低的原因之一在于进口抗抑郁药治疗花费高昂。目的比较选择性5-羟色胺再摄取抑制剂(Selective Serotonin Reuptake Inhibitors, SSRIs) 艾司西酞普兰国产药草酸依地普仑片与专利药来士普治疗抑郁症的有效性和安全性。方法采用随机双盲、阳性药平行对照、多中心临床研究,入组抑郁症病例260例,其中研究组(草酸依地普仑治疗组)和对照组(来士普组)各130例,治疗8周。主要疗效指标为l7项汉密尔顿抑郁量表(Hamilton rating scale for depression, HAMD-17)评分。安全性评估包括不良事件、定期体检、实验室检查和心电图检查等。结果为期8周的治疗中研究组有35名(27%) 受试者脱落,对照组为32名(25%)。意向治疗分析(intention-totreat analysis, ITT)发现治疗8周后,研究组的HAMD量表评分减分(标准差)为13.9(8.2)分,对照组为14.3(8.1)分(t=0.44, p=0.664)。研究组和对照组的有效率(HAMD减分率≥50%)分别为69%和67% (χ2=0.16,df=1, p=0.690);临床痊愈率(研究终点HAMD总分≤7分)分别为51%和49%(χ2=0.06, df=1, p=0.804)。研究组常见的不良反应为口干(12.3%)、恶心(9.2%)和头晕(6.2%),对照组为恶心(10.8%)、乏力(7.7%)和嗜睡(6.9%)。 在研究的前35天中,治疗组出现1例自杀和2例自杀未遂,对照组出现1例自杀(Fisher精确检验,p=0.314)。结论对在精神卫生中心门诊就诊的中、重度抑郁症患者,采用国产草酸依地普仑片与来士普初步治疗的疗效与安全性相当。治疗过程中需要严密关注自杀风险。试验注册号: NCT00866593 (clinical.trails.gov)
Background:Depression is an increasingly important public health problem in China, but only a small minority of patients with this conditi on receive treatment. One of the reasons for low treatment rates is the relatively high cost of imported anti depressant medications.Aim:Compare the efficacy and safety of the generic form of the selective serotonin re-uptake inhibitory (SSRI)anti depressant escitalopram to the proprietary form of escitalopram (Lexapro) in the treatment of major depression.Methods:A multi center double-blinded randomized controlled trial enrolled 260 pati ents with depression and randomly assigned them to receive eight weeks of treatment with either generic escitalopram (n=130) or Lexapro(n=130). Effi cacy was assessed by the Familton rating scale for depression (FAND-17). Safety was assessed by evaluati ng adverse events reported by pati ents, regularly recording vital signs, and conducting laboratory tests and electrocardiograms.Results:There were 35 (27%) dropouts during the 8 weeks of treatment in the generic escitalopram group and 32 (25%) in the Lexapro group. In the intenti on-to-treat analysis (i.e., including all pati ents) the mean (s.d.) drop in the FAND total score at the end of the 8th week of treatment was 13.9 (8.2) in the generic escitalopram group and 14.3 (8.1) in the Lexapro group (t=0.44, p=0.664). The proportions of patients responsive to treatment(i.e., >50% drop in total FAND score) were 69% and 67% in the generic escitalopram group and Lexapro group,respectively (χ2=0.16, df=1, p=0.690; and the proporti ons that achieved remission (i.e., fina FAND <7) were 51% and 49% (χ2=0.06, df=1, p=0.804). The most frequently reported adverse events were dry mouth (12.3%),nausea (9.2 %) and dizziness (6.2%) in the generic escitalopram group and nausea (10.8%), fainting (7.7%) and drowsiness (6.9%) in the Lexapro group. During the first 35 days of treatment, one suicide and two suicide att empts occurred in the generic escitalopram group and one suicide occurred in the Lexapro group (Fisher exact test, p=0.314).Conclusion:Generic escitalopram is as effective and safe as Lexapro in the initial treatment of patients with moderate to severe episodes of major depression who seek treatment in the outpatient departments of psychiatric hospitals in China. Careful monitoring of the risk of suicidal events is an essenti al component of the treatment of depressed pati ents.Trial registration: NCT00866593 (clinical.trails.gov】
10.3969/j.issn.1002-0829.2013.02.007
1Shanghai Nental Fealth Center, Shanghai Jiao Tong University School of Nedicine, Shanghai, China
2Febei Nental Fealth Center, Baoding, Febei Province, China
3The First Affi liated Fospital of Kunming Nedical College, Kunming, Yunnan Province, China
4Xi’an Nental Fealth Center, Xi’an, Shaanxi Province, China
5First Affi liated Fospital of Xi’an, Jiao Tong University Nedical College, Xi’an Shaanxi Province, China
6Xijing Fospital, Forth Nilitary Nedical University, Xi’an, Shaanxi Province, China
*Correspondence: Fuafang Li, lhlh5@yahoo.com.cn; Biao Wang, wangbaio@yahoo.com.cn
(Received: 2012-09-10; Accepted: 2013-01-10)

Dr. Yimin Yu graduated from the Shanghai Second Medical University in 2001 and received a Master's of Medicine degree from the Shanghai Jiao Tong University School of Medicine in 2009.She has worked at the Shanghai Mental Health Center since 2001 and is currently an att ending psychiatrist in the Medical Insti tute for Clinical Trails (MICT) responsible for conducting clinical trials of psychopharmacological agents.
猜你喜欢
江苏安全生产(2022年2期)2022-04-19 13:03:20
汽车工程师(2021年12期)2022-01-18 06:02:43
世界科学技术-中医药现代化(2021年9期)2021-12-31 03:31:36
建材发展导向(2021年23期)2021-03-08 01:05:44
材料科学与工程学报(2016年4期)2017-01-15 13:35:34
信息安全与通信保密(2016年3期)2016-08-23 01:23:46
饮食科学(2016年3期)2016-07-04 15:12:40
饮食科学(2016年3期)2016-07-04 15:12:27
养生保健指南(2016年4期)2016-03-22 12:19:25
电子设计工程(2015年12期)2015-02-27 12:06:24

- 上海精神医学的其它文章
- The stati sti cs of suicide
- Case Report of Rapid-eye-movement (REM】 sleep behavior disorder
- Mild cogniti ve impairment: a concept useful for early detection and intervention of dementia
- Pesticides availability and medically serious suicide attempts in China
- Relationship of changes in cognitive and depressive symptoms during anti depressant treatment of individuals with geriatric depression and their relationship to the APOE epsilon 4 allele
- Methodology of China's nati onal study on the evaluati on,early recogniti on, and treatment of psychological problems in the elderly: the China Longitudinal Aging Study (CLAS】