
高血糖对树鼩脑皮层局灶性缺血时海马微环境离子稳态及神经元继发性损伤的影响*
2013-12-01 02:28李树清
中国病理生理杂志 2013年8期
陈 静, 李树清
(昆明医科大学病理生理学教研室,云南昆明650500)
缺血性脑血管病是人类致死致残的主要疾病之一。许多因素都可诱发或加重脑缺血的病理过程,其中,高血糖和以血糖增高为主要特征的代谢性疾病——糖尿病就是缺血性卒中的高危因素之一。高血糖可以通过多种途径和机制对缺血后神经元损伤发挥影响[1-2]。近年来,缺血后神经元微环境变化与神经元损伤的关系备受关注。神经元微环境广义上说,是神经元所处的局部理化环境,包括星形胶质细胞、基质细胞、微循环以及主要由细胞外基质和组织间液所构成的细胞外间隙[3]。研究发现,缺血后脑损伤的一系列病理生理变化中,局部酸化是组织缺血常见的共有特征,而钙离子超载和细胞内水钠潴留又是脑损害的关键环节[4-5]。因此,神经元微环境中酸碱平衡以及离子稳态的维持对于缺血后神经元的生存至关重要。本实验选择对缺血敏感的海马区作为主要观察对象,研究脑皮层局灶性缺血及高血糖条件下,离子微环境变化与海马神经元继发性损伤间的相互关系,探讨高血糖在缺血性脑损伤中的作用及其机制,从而为控制卒中危险因素、保护易受损神经元提供参考。
材料和方法
1 材料
1.1 动物与分组 健康成年树鼩40只,雌雄不拘,体重(120±20)g(由昆明医学院实验动物中心提供)。随机分为正常血糖(normoglycemia,NG)和高血糖(hyperglycemia,HG)2个大组,每个大组又随机分为假手术组、缺血4 h、缺血24 h及缺血72 h 4个亚组,即有:正常血糖假手术组(NG-sham)、缺血(ischemia,Is)4 h组(Is4)、缺血24 h组(Is24)、缺血72 h组(Is72)、高血糖假手术组(HG-sham)、HG+Is4组、HG+Is24组和HG+Is72组,共8个亚组。每个亚组均为5只动物。安静、避光、相同条件下饲养。
1.2 试剂 链脲佐菌素(streptozocin,STZ),Sigma产品;孟加拉红(rose bengal),Fluka产品,以0.85%生理盐水溶解,浓度为15 g/L;林格氏液,四川科伦药业股份有限公司产品,临用前调定pH值至7.4使用。
1.3 器材 SQ-Ⅲ型光化学诱导脑血栓形成装置(本室研制);随手测血糖仪及血糖试纸(LifeScan产品);单泵等速推挽式微灌流系统(本室研制);KW-Ⅱ型脑立体定位仪(西北光学仪器厂产品);DH-505型K+/Na+/Cl-/Ca2+/pH电解质分析仪(南京安科电子医疗设备有限公司生产);PHS-2C精密酸度计(上海雷磁仪器厂生产)。
2 方法
2.1 STZ诱导树鼩实验性高血糖模型的制备[6]树鼩禁食12 h,不禁水,采集手掌末梢血测定空腹血糖(fasting blood glucose,FBG)。高血糖组腹腔1次性注射新鲜配制的 STZ溶液(STZ临用前以0.1 mol/L、pH 4.2的无菌柠檬酸盐缓冲液溶解配制成2%STZ溶液),剂量为120 mg/kg;正常血糖组仅腹腔注射同等剂量的缓冲液。所有动物给予同样标准的饲料和充足的水分,给药3 d以后再次采血测定FBG,将FBG≥11.1 mmol/L者列为高血糖组实验对象。
2.2 树鼩皮层血栓性脑缺血模型的建立[7]以1%硫喷妥钠溶液,按5 mL/kg腹腔注射麻醉树鼩,俯卧位固定。于右侧颞顶部作约1.5 cm长的弧形切口,分离皮下组织,暴露颅骨。于切口处皮下嵌入一外径2.5 cm、中心处有直径0.5 cm圆孔的消毒铝片,铝片周围用避光纸遮盖保护。至此,经舌下静脉一次性注射孟加拉红生理盐水溶液1.33 mL/kg,循环10 min后,将动物置于SQ-Ⅲ型光化学诱导脑血栓形成装置下,在光强度1.0 W/cm2下照射15 min,照射期间颅骨表面温度维持在(36.5±1.0)℃左右。照毕,取出铝片、缝合切口、保温待动物清醒后放回饲养笼内观察。假手术组也按规定剂量注射孟加拉红而不予光照。
2.3 树鼩海马微灌流及离子微环境检测 实验组分别于光化学反应后4、24及72 h,假手术组于光化学反应后24 h时点,提前1.5 h麻醉动物,固定在立体定位仪上。以法兰克福平面(Frankfurt plane)作为立体定位坐标系统的基础,这一平面通过外耳道的中心以及眶口的下缘,冠状面与法兰克福平面垂直并同时通过两外耳道的中心,形成AP 0.0平面[8]。选择右侧海马为灌流部位,以AP 0.0平面前2.5 mm(A 2.5)、中线右侧6.5 mm(R 6.5)坐标点处为穿刺点,在颅骨上钻直径为1 mm的小孔,勿破坏硬脑膜。以电极固定器夹持微灌流探针,在数字式微电极驱动器控制下,自硬脑膜表面向下垂直进针8.0 mm(H 8.0)至海马,置入微灌流探针后,用单泵等速推挽式微灌流系统以林格氏液为灌流液,按10μL/min的灌流速度分别对各组动物行海马微灌流,维持灌流液温度为37℃,平衡预灌流60 min后,收集灌流液标本0.5 mL,避免与空气接触。所得标本立即用离子分析仪进行 Na+、K+、Ca2+、Cl-含量及 pH 值分析。
2.4 脑组织形态学观察
2.4.1 脑组织固定与取材 动物仰卧位固定,暴露心脏,钳闭降主动脉,直视下将灌注管经左心室进入升主动脉。剪开右心耳,先用4℃的0.85%生理盐水100 mL冲洗,再用4℃的10%中性缓冲甲醛200 mL灌流固定,动物僵直为满意。取出整脑,用10%中性缓冲甲醛后固定48~72 h。冠状切取视交叉后1~4 mm的脑组织,常规脱水、石蜡包埋,由前向后连续行冠状切片,片厚4μm。
2.4.2 HE染色 将石蜡切片脱蜡至水,HE染色,梯度乙醇脱水,二甲苯透明,中性树脂封片后显微镜观察。
2.4.3 形态学分析 显微镜下观察缺血侧大脑皮层缺血中心区和半暗带的形态学改变。同时,选择对缺血敏感的海马CA1区进行脑损伤定量评价。(1)组织学分级:在光学显微镜下以相同光亮度对海马CA1区组织学改变进行分级,标准如下:0级,无神经元死亡;Ⅰ级,散在的神经元死亡;Ⅱ级,成片的神经元死亡;Ⅲ级,神经元几乎全部死亡。(2)存活神经元计数:因海马CA1区呈带状分布,且为了排除坏死、凋亡及各种变性细胞的区别所造成的影响,所以只计数正常的存活细胞数,即用微计数尺在20倍物镜下计数患侧海马CA1区每毫米的存活神经元数,每张切片计数3个区段,取其平均数为神经元密度(neuronal density,ND),作为评价缺血损害的指标。
3 统计学处理
数据以均数±标准差(mean±SD)表示,用SPSS 11.5统计软件包,行正态性检验t检验、方差齐性检验、One-way方差分析以及样本均数间的两两比较(LSD法),以P<0.05为差异有统计学意义。
结 果
1 给予STZ前后树鼩血糖水平的变化
腹腔注射缓冲液的树鼩,血糖水平在给药前后无明显差异;而腹腔注射120 mg/kgSTZ,能使树鼩血糖水平显著升高(P<0.01),制备出符合要求的实验性高血糖动物模型,见表1。

表1 树鼩空腹血糖的变化Table 1.The changes of fasting blood glucose in tree shrews(mmol/L.Mean±SD.n=20)
2 脑缺血及高血糖脑缺血条件下海马离子微环境的变化
缺血组动物缺血后各时点pH值与假手术组相比均有下降,以缺血后4 h最为显著(P<0.01),其次为缺血后24 h(P<0.05);K+含量在缺血后4 h明显升高(P<0.01),其余时点的变化与假手术组相比无显著差异;Na+在缺血后4 h(P<0.01)和24 h(P<0.05)明显下降;Ca2+在缺血后4 h降低(P<0.01);Cl-同样也在缺血后 4 h降低(P <0.05),其余时点无明显变化。单纯高血糖组树鼩,海马微环境中 pH 值、K+、Na+、Ca2+和 Cl-的含量与正常血糖假手术组相比均无明显差异(P>0.05)。高血糖加缺血组动物缺血后各时点pH值、K+、Na+、Ca2+和Cl-含量的变化规律与正常血糖缺血组相似:K+含量在缺血72 h后也有升高(P<0.05);Ca2+含量在缺血24 h后依然降低(P<0.05);Cl-在缺血后的各时点均未见明显变化(P>0.05)。缺血后4 h,高血糖组pH值、K+和Ca2+含量比正常血糖组变化更为明显(P<0.05);而缺血后24 h,高血糖组的 pH值及Na+含量与正常血糖组同期值相比,下降也更为显著(P <0.05),见表2。
3 脑缺血及高血糖脑缺血条件下的脑组织形态学分析
HE染色显示,NG-sham组皮层结构完整,海马形态结构正常,锥体细胞排列紧密整齐,见图1A。光化学反应后4 h即可见缺血性损伤改变。24 h病变尤为明显,皮层缺血中心区液化、坏死,神经元脱失;半暗带皮质水肿,神经元肿胀、固缩;海马CA1区锥体细胞稀疏、排列散乱、出现典型的缺血损伤性改变,胞体缩小呈三角形、胞核固缩深染、核仁消失,见图1B。缺血后72 h,仍可见到缺血固缩的神经元,并伴随胶质细胞增生等修复性反应。HG-sham组树鼩皮层及海马存在散在的细胞损伤性改变,细胞周围空晕形成,胞质浓染,胞体变小,见图1C。HG+Is组各时点组织学特征与Is组相似,但损伤情况更为严重,皮层出现较大液化灶,海马CA1区锥体细胞层次不清,成片死亡,见图1D。
从各组动物患侧海马CA1区组织学分级中可以发现,光化学诱导树鼩脑皮层缺血后4 h,在同侧海马区也检测到了海马神经元的继发性损伤,24 h达高峰,72 h损伤仍很明显;高血糖本身也会引起神经细胞损伤;高血糖加缺血组在各时点的CA1区组织学分级更高,海马神经元继发性损伤更为严重,见表3。
海马CA1区存活神经元计数显示,NG-sham组ND 为188.00±15.95;Is组 ND 下降,缺血后4 h、24 h和72 h 的 ND 分别为 106.60 ±19.81、78.40 ±15.31和95.80±13.01,与 NG-sham 组相比,均 P <0.01;升高血糖也会引起神经元损伤,HG-sham组ND为 160.40 ±18.41,与 NG-sham 组相比,P <0.05;HG+Is组的缺血性损伤加重,在缺血后4 h、24 h和72 h 的 ND 值分别为 92.80 ±20.10、42.00 ±17.29 和72.20 ±17.91,与 Is组的同期值相比,在缺血后24 h和72 h差异尤为明显(P<0.05),见图2。
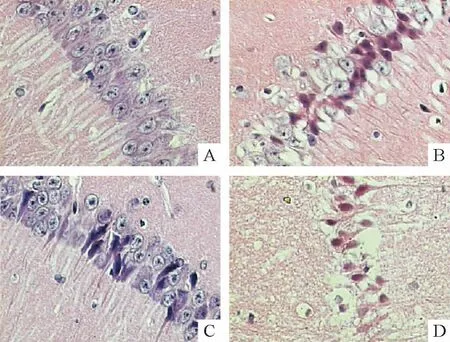
Figure 1.The effects of focal cortical ischemia and hyperglycemia on the histomorphology of the ipsilateral hippocampus in tree shrews(HE staining,×200).A:normoglycemia(NG)-sham group;B:24 h after ischemia in NG group;C:hyperglycemia(HG)-sham group;D:24 h after ischemia in HG group.图1 脑皮层局灶性缺血及高血糖对树鼩同侧海马组织形态学的影响
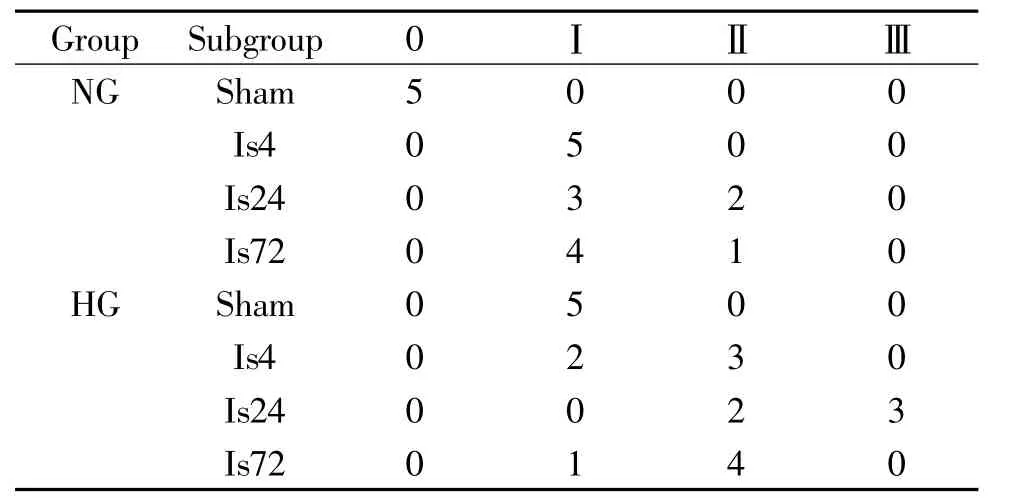
表3 树鼩脑皮层缺血后患侧海马CA1区组织学分级的变化Table 3.Changes in the histological grades of the ipsilateral hippocampal CA1 area after cortical ischemia in tree shrews(n=5)
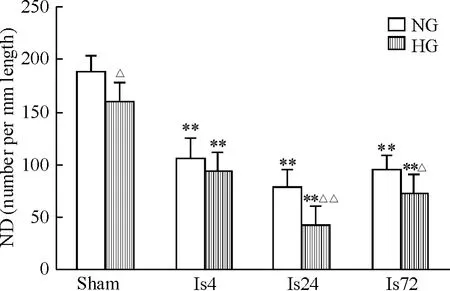
Figure 2.Changes of the intact pyramidal neuronal density(ND)in the hippocampal CA1 area after cortical ischemia in tree shrews.NG:normoglycemia;HG:hyperglycemia;Is4,Is24 and Is72:4,24 and 72 h after ischemia,respectively.Mean ± SD.n=5.*P <0.05,**P <0.01 vs sham;△P < 0.05,△△P < 0.01 vs NG.图2 树鼩脑皮层缺血后患侧海马CA1区存活神经元密度的变化
讨 论
神经元是构成神经系统结构和功能的基本单位,其生活在特定的微环境中;神经元微环境是一个成分多样、组成复杂的细胞生存空间。对神经元微环境的探讨是近年来神经科学研究的热点之一。在微环境的组成中,细胞与细胞、细胞与细胞外间隙以及细胞外间隙的不同要素之间均存在密切的相互作用和制约,这种复杂的网络联系发挥着微妙而精细的调节功能,促成神经元微环境性质的稳定。缺血、缺氧、pH值、酸碱、血糖水平等的改变,均可引起微环境的改变,导致细胞凋亡或坏死的发生。
本实验利用光化学方法诱导血栓形成,从而在树鼩右侧顶叶皮层复制出局灶性脑缺血模型。我们不仅可以在缺血侧皮层观察到明显的梗死灶,而且在患侧海马也检测到神经元的继发性损伤及其离子微环境(细胞外 pH 值、K+、Na+、Ca2+、Cl-)的显著改变。同时,微环境内酸碱平衡以及离子稳态性的破坏恰恰也就为组织形态学结果提供了合理解释及有力证明,我们认为海马离子微环境的改变可能是造成海马细胞继发性损伤的重要原因。那么,皮层缺血为什么会引起海马微环境的紊乱呢?研究显示,海马神经元对脑缺血非常敏感、具有高度选择性易受损的特点[9];并且,由于海马组织与脑皮层及侧脑室位置相毗邻;其传入纤维丰富、与周围组织密切联系,这就使得海马细胞微环境易于受到缺血中心区的影响。因此,我们推测,海马微环境的改变与皮层缺血中心区有关,可能是皮层缺血区缺血微环境恶化并向周围扩散的结果。海马微环境受损的机制可能涉及以下几方面:(1)缺血区去极化及皮层扩布性抑制(spreading depression,SD)。缺血区去极化是由梗塞中心缺氧性K+释放及缺氧性谷氨酸释放所触发的。而SD是指大脑皮层神经元在持续有害刺激下,出现的一种伴有细胞膜去极化的、缓慢、扩布性的、短暂、可逆的皮层电生理活动的抑制[10]。SD是一种短暂的离子波,包括有K+的释放和Ca2+、Na+及Cl-的吸收[11]。本研究中离子微环境的变化趋势也与之相符合。由于离子浓度的变化,导致兴奋性氨基酸释放增加,引起梗塞灶周围出现重复的病理性SD,使缺血性损害加大。因而我们推测海马神经元离子微环境平衡的破坏可能是SD的特殊表现之一。(2)海马微环境的改变与海马区血管受损及血流动力学异常有关。本实验中,脑缺血的形成是以光化学反应产生单线态氧,使照射部位血管内皮细胞受损作为始发因素的。血管内皮细胞受损后可进一步激活凝血因子、内源性血小板活化因子产生增加、释放大量的活性氧、自由基及炎症介质等。这些异常增多的血管活性物质不仅可以引起光照局部血栓形成,发生缺血梗死;而且它们尚可通过血液循环、细胞外间隙、神经元网络联系等途径转移、扩散至其它区域,如与之毗邻且对缺血易感的海马,进而使海马区血管受损和血小板活化,诱导血栓形成,从而直接造成海马区缺血微环境的形成。形态学检查中,我们在缺血侧海马区小血管可见到微血栓的形成,就给予上述推测以有力支持。
在本研究中,高血糖不仅是缺血性脑血管病的危险因素之一,也是缺血性脑损伤的恶化因素。与正常血糖缺血组同期值相比,高血糖加缺血组动物的损伤程度和范围均有所加重,皮层梗死灶更大,海马CA1区组织学分级更高、存活神经元密度更低;而这又与高血糖加重细胞外酸中毒、加剧缺血微环境的紊乱密切相关。光化学诱导皮层脑缺血后引起海马细胞微环境内pH值的降低,变化以缺血后4 h最显著,而高血糖组在缺血后4及24 h时,细胞外液pH值下降程度则更为明显。这是因为脑缺血时葡萄糖无氧酵解加速,乳酸生成增多,且此时能量匮乏,膜转运功能失常,对酸碱平衡进行缓冲和调节的能力亦降低,致使细胞内外pH下降。而高血糖状态增加了底物的供给,乳酸大量堆积,加之缺血组织脑循环受阻,使乳酸积累到更高水平,从而加重酸中毒。同时,在高血糖状态下,由于严重的酸中毒及能量匮乏等原因,使细胞膜通透性增加及离子泵活性降低,进一步增加了 Na+、Ca2+内流和 K+的外流。此外,高血糖也抑制胶质细胞的活化[12]。星形胶质细胞富含糖元,其pH值下降程度比其它细胞更重,更易发生变性、坏死,从而丧失对神经细胞的营养支持作用,也丧失对细胞外液离子稳态性的调节能力,更加重了细胞内钙超载和钠水潴留。而能量代谢的变化以及微环境离子稳态性的紊乱可进一步通过加速自由基的产生,增强兴奋性氨基酸的毒性作用,干扰细胞内的信号转导和蛋白质合成等分子机制,激发引起细胞快速死亡的级联反应,造成脑细胞的继发性损伤。由此可见,海马微环境的改变可能是造成海马神经元继发性损伤的重要原因之一,高血糖加重缺血性脑损伤的作用与其加剧缺血微环境的紊乱互为因果,形成恶性循环,从而使得对正常血糖者而言是可逆性的时间窗对高血糖或糖尿病患者而言变得不可逆。
猜你喜欢
中西医结合心脑血管病杂志(2022年18期)2022-10-21
中国医药科学(2022年5期)2022-05-05
载人航天(2021年5期)2021-11-20
天津医科大学学报(2021年3期)2021-07-21
自我保健(2021年4期)2021-06-16
中国医学影像技术(2020年11期)2021-01-04
中华养生保健(2020年7期)2020-11-16
中国现代医药杂志(2020年3期)2020-05-08
中国生物医学工程学报(2019年6期)2019-07-16
中国当代医药(2015年10期)2015-03-01
