
多肽和蛋白质药物口服吸收机制及策略的研究进展
2013-11-12 07:24:24张文平杨久春吕正兵
中国药理学与毒理学杂志 2013年5期
张文平,杨久春,吕正兵,陶 雷,陈 洁
(1.浙江理工大学生命科学学院生物化学所,浙江杭州 310018;2.黑龙江迪龙制药有限公司,黑龙江安达 151400)
随着生物技术的发展,很多活性多肽和蛋白质被开发成药物并应用于临床。与化学药物相比,多肽和蛋白质药物(peptide and protein drugs,PPD)具有作用靶点专一、不良反应少以及药效作用强等优点,故在现代疾病的预防和治疗中的应用日益广泛;但多肽和蛋白质由于分子量大、结构相对复杂和理化性质特殊,机体对其产生了多种屏障,如酸屏障、酶屏障和膜屏障,限制了这类药物的口服吸收[1-2]。目前,上市的PPD常规给药途径多以注射为主。PPD口服给药是一种方便且提高患者依从性的给药方式,自20世纪90年代以来,国内外对这一领域的研究非常活跃。克服PPD的口服给药技术难题,将会给生物制药工业带来巨大变化并造福人类。本文从PPD口服的认识过程、吸收机制、影响因素及提高策略等方面进行了综述。
1 多肽和蛋白质药物认识过程
随着现代生物技术迅猛发展,多肽或蛋白质不断被开发成药品,以红细胞生成素、干扰素和胰岛素为代表的近百种PPD已经在临床应用。1982年,世界第一个基因工程产品人胰岛素上市;1992年,我国第一个基因工程产品干扰素α1b上市[1]。但上述PPD在临床上只有通过im,iv或sc给药才能产生治疗作用,药物学家一直致力于PPD口服制剂研究。
关于PPD口服吸收,过去人们一直认为蛋白质经胃肠道酶降解后以氨基酸形式被吸收,后来发现其消化产物主要是多肽如二肽和三肽等。20世纪90年代,对PPD口服吸收有了进一步的认识,PPD可通过液相或受体介导的胞饮作用方式吸收入血,对于12个以下氨基酸组成的多肽,实现口服给药的可能性很大,而对于蛋白质,则难以预料。但是,多学科的合作为PPD口服制剂的成功开发提供了机遇。
进入21世纪,对PPD口服吸收机制又有了新的认识,发现一些多肽可通过肠多肽转运系统吸收入血,拓展了PPD吸收途径[3]。逐渐意识到要成功开发PPD口服制剂,必须解决酶屏障和膜屏障两个关键问题[4],利用纳米材料、脂质体和酶抑制剂等技术,完全可能在不久的将来攻克这一难题[5-7]。
2 多肽和蛋白质药物口服吸收的机制及途径
2.1 载体转运
小分子药物的转运以简单扩散为主,而PPD口服给药经过胃肠道主要依靠载体转运介导通过细胞旁路转运至小肠黏膜内[8],如图1所示,随后PPD由淋巴回流进入血液循环系统。未被消化酶降解的多肽与肠表面膜基底外侧的H+依赖型多肽载体结合,以H+梯度和膜电位差为动力,经多肽载体转运进入基底膜内侧,由于H+与多肽是共同通过上皮细胞膜的,这一系统又称为H+-依赖型肠多肽转运系统[3]。
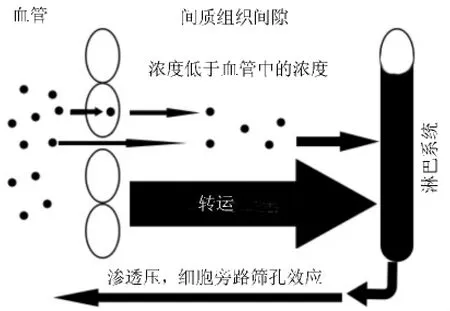
图1 治疗用多肽与蛋白质药物分布机制:载体转运的作用超过简单扩散[8].
2.2 胞饮作用和M细胞途径
当PPD与小肠黏膜刷状缘膜的受体结合后,结合部位膜内陷并形成吞噬体而进入小肠壁(图2),可将40 ku大小的完整蛋白质转运至细胞内[9],但有很少一部分PPD通过胞饮作用从血管外转运到小肠上皮细胞内。

图2 多肽和蛋白质药物分子通过胞饮从血管外进入小肠细胞[9].
PPD通过胞饮作用进入小肠上皮细胞,最典型的例子就是通过M细胞途径(图3),M细胞是一种位于肠集合淋巴结上的微皱褶细胞,含有M细胞小囊和派伊尔淋巴集结[10],M细胞表面能吸附颗粒,它可将完整的抗原蛋白或病毒分子经胞吞、胞吐途径吸收并呈递给黏膜下免疫细胞[3]。
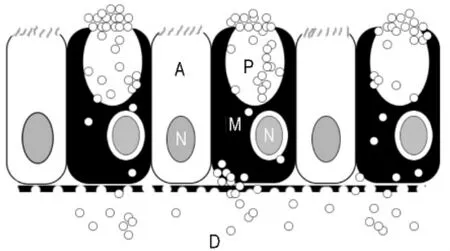
图3 肠淋巴微皱褶细胞(M细胞)吞噬颗粒的原理图.相互间隔的M细胞与柱细胞(A)都含有细胞核(N)。位于M细胞表面的颗粒(P)→M细胞的囊泡→派伊尔淋巴集结(D)[10].
2.3 细胞旁路
如图4所示,PPD可以经过细胞旁路被动转运,不需要消耗能量,依靠肠上皮细胞两侧的药物浓度、电化学势的差别形成的。阻碍PPD经细胞旁路入血的是小肠上皮细胞之间的紧密连接或闭合带,但是,餐后在肠腔葡萄糖刺激下,肠黏膜细胞骨架收缩,紧密连接暂时开放,某些相对分子质量为0.9~1.9 ku的 PPD 可经此途径吸收入血[11]。

图4 多肽和蛋白质药物通过细胞旁路(A)和跨细胞途径(B)进入血液[11].
3 影响多肽和蛋白质药物口服吸收的因素
3.1 相对分子质量
由于PPD相对分子质量大,人的小肠黏膜细胞间隙为5 nm,一般情况下难以透过小肠上皮以简单扩散的方式转运入血。小鼠的肠黏膜孔径约0.4 nm,氨基酸、二肽和三肽可以穿过肠壁,多肽和蛋白质则难以通过[1]。
3.2 空间结构
影响PPD活性的因素主要是氨基酸序列、末端基团、肽链和二硫键及其空间结构。蛋白质和多肽的三级结构在体外穿过人结肠癌细胞的分子层中发挥了重要作用,这是由于蛋白质的部分展开,使疏水部分暴露,可以穿过液态脂质双分子层。
3.3 理化性质
蛋白质是一种亲水性高分子化合物,决定了它既不能通过细胞间隙简单扩散,也难以与细胞膜融合,如果不借助载体则很难通过小肠黏膜入血,因此生物利用度较低[1]。
3.4 酸屏障、酶屏障和膜屏障
(1)酸屏障 由于大多数多肽和蛋白质不耐酸,胃酸的pH值为1~3,当PPD通过胃时,一部分药物被水解失去生物活性,而且整个肠道pH环境复杂多变,一些多肽还容易发生pH依赖性水解[2,12],这成为PPD口服吸收的第一道屏障难以逾越。
(2)酶屏障 胃肠道含有,如多种消化酶,主要包括:①腔道分泌的酶,如胃蛋白酶、胰蛋白酶和糜蛋白酶等;② 刷状缘分泌的酶,如羧肽酶、氨肽酶、肽链内切酶和外切酶等,酶消化作用是导致PPD降解的主要原因[13],这是PPD口服吸收的第二道屏障。
(3)膜屏障 首先小肠的上皮细胞附着一层水合凝胶,称为不流动水层,其下方是磷脂表面活性层,疏水性强,阻碍了PPD等亲水性分子向上皮细胞的扩散。其次,小肠上皮细胞膜的磷脂双分子层具有半透膜性质,允许脂溶性药物被动转运,而PPD等大分子以及亲水性药物需要通过主动转运才能跨膜进入细胞内[14]。最后,阻止PPD口服吸收的主要屏障是致密的肠上皮细胞膜,因为小肠细胞间隙仅为5 nm,在不加任何促吸收剂的情况下,只有分子量小的药物才能通过。总之,小肠黏膜特殊的组成和结构是PPD口服吸收的第三道屏障[2]。
3.5 首过效应
有些从胃肠道吸收入门静脉系统的药物进入肝后被肝药物代谢酶代谢,具有首过效应。肠道的刷状缘、肠内皮细胞溶酶体同样参与首过效应。胆汁内容物也有一定的作用,包括水解、氧化、脱烷基化和还原等。一部分药物被这些酶代谢,从而使药物进入血液循环的有效量减少,故首过效应是导致PPD口服生物利用度低的原因之一[1]。
4 提高多肽和蛋白质药物口服吸收的策略
4.1 化学修饰
通过对PPD进行化学修饰形成前药或衍生物,可以保护PPD免受蛋白酶等消化酶的降解作用,对PPD的结构修饰一般要考虑亲脂性、电荷数、等电点、化学稳定性以及对载体的亲和力等[2]。例如,酯化反应可将多肽或蛋白质的疏水基团以共价键或非共价键结合起来,从而提高PPD的亲脂性,但酯化反应可能会降低PPD的生物活性,最近研发的一种可逆酯化反应技术可保证酯化的PPD经口服后再生恢复其生物活性,已有几种PPD经过这种技术改造后,提高其在胃肠道的吸收程度、稳定性及生物活性。聚乙二醇共轭修饰不仅提高PPD脂溶性并且有防止酶解的作用。美国Nobex公司通过亲水性聚乙二醇链和亲脂性烷基链在赖氨酸-29位点上对人重组胰岛素进行共价修饰,临床试验证明可以治疗1型和2型糖尿病,生物利用度为5%[15]。
4.2 吸收促进剂
吸收促进剂是一类可逆性增强药物胃肠道透过性的小分子化合物,是提高生物膜通透性的主要方法。吸收促进剂可以非特异性地暂时打破小肠屏障、改变细胞紧密连接的完整性及扩大细胞间隙[16],或者通过干扰膜磷脂双分子层稳定性使其形成小孔,从而提高生物膜通透性,增强PPD等药物经胃肠道吸收入血。传统的吸收促进剂如胆酸盐类、表面活性剂、水杨酸类、氨基酸类衍生物和金属螯合剂等长期使用后具有一定的不良反应,制约其应用。
近年研制的一些不良反应小的新型吸收促进剂,如细胞膜穿透肽,可以介导药物分子跨膜进入不同组织和细胞,其富含碱性氨基酸的一级结构和螺旋结构对穿膜起着重要作用,日本学者研制基于精氨酸残基组成的细胞膜穿透肽促进了胰岛素经鼻黏膜、口服给药的吸收[17-18]。Petrus等[4]用维生素B12与胰岛素制成纳米粒,有效地促进胰岛素口服吸收。
4.3 酶抑制剂
酶抑制剂能够有效地防止酶对PPD的降解,研究表明PPD与酶抑制剂联合应用后,其口服生物利用度显著提高[2]。
酶抑制剂可分为4类:① 非氨基酸类 P-氨基苯甲酰胺、1,2,3,4-四氢-1-萘甲酸 4-(4-异丙基-1-哌啶羟基)-苯酯甲烷磺醋(FK-448)和甲磺酸卡莫司他(camostat mesylate);②氨基酸类 肽类和修饰肽;③胰凝乳蛋白酶抑制剂、氨肽酶抑制剂、杆菌肽和镇痛素;④ 肽酶抑制剂 抑肽酶和胰蛋白酶抑制剂[13,16]。应用包曼-伯克抑制剂和弹性蛋白酶抑制剂制备壳聚糖降钙素口服制剂,可以降低其在胃肠道内的降解[19]。
4.4 纳米粒载体
纳米粒是直径在10~100 nm的超微小药物载体,可通过细胞旁路或肠黏膜派伊尔淋巴集结的M细胞吞噬途径吸收入血[1],纳米粒包裹PPD可以避免胃肠道分泌的酸和消化酶的破坏,载体纳米粒粒径越小,越易吸收,且具有一定靶向性。
制备纳米粒的载体材料可分为两种类型:生物降解型和非生物降解型。有机体内的水解酶可以将生物降解聚合物材料水解成水和二氧化碳,近年来对生物降解聚合物材料的研究更为重视,常见的生物降解型纳米材料有交链聚酯、聚原酸酯、聚酐和多肽等[20]。以研究胰岛素口服制剂等为代表的载体纳米微粒见表1,其中壳聚糖纳米粒在开发口服胰岛素用于治疗糖尿病取得了一定的成效[12]。
4.5 脂质体载体
脂质体的结构与细胞膜双层脂膜结构类似,可通过肠黏膜细胞的吸附、脂质交换、融合和内吞等作用机制,增强细胞摄取药物,特别是促进了派伊尔淋巴集结对药物的摄取。而且,脂质体保护PPD等药物免受胃肠道酶的降解,起到缓释作用,延长半衰期[7]。Kv 等[22]等制备沙雷菌肽酶(serrationpeptidase)脂质体并以Caco-2细胞模型研究发现,脂质体剂型比原药有更好的跨膜转运效果。Hanato等[23]制备的胰高血糖素样多肽脂质体在动物实验中具有良好的降血糖作用。但是,脂质体用于PPD载体存在两个问题:①脂质体在制备过程中,PPD的包封率较低;② 脂质体的不稳定性,因为胃肠道存在的胆盐会破坏脂质体的双分子层结构,使脂质流失,导致脂质体的稳定性比较差[24]。
4.6 微乳载药
乳剂中的油相可增加PPD的膜通透性、也可能增加药物的淋巴转运,从而提高生物利用度[1]。早在1997年Cortecs公司就开发了胰岛素油制剂,通过两亲性表面活性剂的作用,将亲水性药物如胰岛素、降钙素等增溶至弱极性的油中形成澄清透明的溶液,6例健康志愿者口服该制剂后血糖降低34%[25]。国外已有环孢素微乳软胶囊上市,国内也将胰岛素制成复乳,ig给予小鼠后有显著降血糖作用[1],北京大学研制的蚓激酶油包水口服微乳具有较好的可行性[26]。
4.7 定位释药
胃肠道存在PPD特异性吸收的位点,如果使PPD在此释放,可避免各种消化酶的降解作用,最大程度地提高PPD的口服吸收,一般从结肠和小肠考虑定位释药。PPD吸收的最佳部位可能是大肠[1],因为药物在此停留20~30 h,而且消化酶的活性较低。研究较多的是结肠定位释药,它具有延长药物的滞留时间、降低酶活性和增强吸收促进剂的作用等优势,常用α-淀粉、果胶钙、壳聚糖、海藻酸、丙烯酸酯偶氮苯水凝胶以及果胶/半乳甘露聚糖等作为包衣材料[27]。小肠的某些区域存在着与免疫有关的特定组织区域,如派伊尔淋巴集结能使淋巴因子和一些颗粒进入循环系统,使用肠溶衣或其他控释技术使药物在小肠释放,可能增加PPD的吸收。

表1 口服载药纳米颗粒[21]
4.8 基因工程
Page等[28]认为基因工程在PPD口服制剂开发方面具有很大潜力,口服基因药物递送载体包括病毒载体和合成高分子载体等,如霍乱毒素B亚单位对GM1神经节苷脂(细胞膜糖脂类受体)有高度亲和力,当霍乱毒素B亚单位连接到包裹抗原的脂质体表面时,小鼠口服给药后,可显著升高黏膜免疫球蛋白A和免疫球蛋白G水平。有人将热休克蛋白A和霍乱弧菌封闭小带毒素两个基因进行融合,并在大肠杆菌中高效表达,研制成幽门螺杆菌热休克蛋白A-霍乱弧菌封闭小带毒素融合蛋白质口服疫苗[1]。
本实验室采用家蚕杆状病毒表达系统对PPD口服给药做了系列研究,利用基因工程改造功能基因并在家蚕杆状病毒表达系统表达一些用于口服的重组蛋白如霍乱毒素B亚单位-重组人胰岛素、重组人粒细胞-巨噬细胞集落刺激因子、重组人乳铁蛋白、重组人生长激素、重组人破骨细胞形成抑制因子和口服基因工程疫苗等[29-33]。Gong等[31]构建霍乱毒素B亚单位人胰岛素融合基因,利用家蚕杆状病毒高效表达霍乱毒素B亚单位-重组人胰岛素融合蛋白,ig给予非肥胖型糖尿病小鼠后抑制了胰岛炎症和糖尿病病理症状的发生;Meng等[34]在家蚕杆状病毒表达系统里成功表达了霍乱毒素B亚单位-重组人胰岛素-绿色荧光蛋白融合蛋白,对该融合蛋白经小肠黏膜转运过程进行追踪,发现其ig给予10周龄的非肥胖型糖尿病小鼠后,产生了特异性抗体,并且诱导CD4+CD25+Foxp3+T细胞的产生,延缓1型糖尿病的发作,改善其症状。目前,这项利用家蚕生物反应器生产的胰岛素口服制剂是一种治疗1型糖尿病的新型药物,现处于中试阶段。家蚕生物反应器表达的重组人粒细胞-巨噬细胞集落刺激因子,ig给予小鼠、Beagle犬和猕猴后,具有升高白细胞作用;健康志愿者口服后其生物利用度平均为1%[29],现处于Ⅱ期临床研究阶段。Liu等[33]在家蚕细胞中表达重组人乳铁蛋白,表达量约为13.5 mg·L-1,小鼠ig给药后,可有效缓解葡聚糖硫酸钠诱导的小鼠急性结肠炎症状。
5 展望
自从1982年世界第一个基因工程产品人胰岛素问世以来,大量的PPD就被开发并应用于临床。2010年全球最畅销、销售额达到50亿美元以上的12种药品中,有6种为生物制剂。截止2011年6月,美国食品药品监督管理局已批准包括重组细胞因子、融合蛋白和基因工程疫苗等各类上市生物技术药物39种,并有超过120种后选药物处于不同临床研究阶段,尽管口服PPD所占比例很少,但是已经取得很大进展。由于PPD口服制剂产品技术含量及附加值高,患者耐受性好,因此其应用前景广泛,国内外很多制药公司、研究机构正加快对PPD口服制剂的研发,PPD口服制剂已成为医药工业发展的重要方向之一,也是新药研发的重点和难点。相信经过多学科的合作,人类最终会成功开发PPD口服制剂。
[1]Zhang WP.PhaseⅠ clinical trial of BmrhGM-CSF and absorption mechanism of BmrhGM-CSF by oral administration(BmrhGM-CSF的Ⅰ期临床试验及其口服吸收机制)[D].Hangzhou:Zhejiang University,2010.
[2]Shaji J,Patole V.Protein and peptide drug delivery:oral approaches[J].Indian J Pharm Sci,2008,70(3):269-277.
[3]Toorisaka E,Watanabe K,Ono H,Hirata M,Kamiya N,Goto M.Intestinal patches with an immobilized solid-in-oil formulation for oral protein delivery[J].Acta Biomater,2012,8(2):653-658.
[4]Petrus AK,Fairchild TJ,Doyle RP.Traveling the vitamin B12pathway:oral delivery of protein and peptide drugs[J].Angew Chem Int Ed Engl,2009,48(6):1022-1028.
[5]Shen WC.Oral peptide and protein delivery:unfulfilled promises?[J].Drug Discov Today,2003,8(14):607-608.
[6]Wang XQ,Zhang Q.pH-sensitive polymeric nanoparticles to improve oral bioavailability of peptide/protein drugs and poorly water-soluble drugs[J].Eur J Pharm Biopharm,2012,82(2):219-229.
[7]Griffin BT,O'Driscoll CM.Opportunities and challenges for oral delivery of hydrophobic versus hydrophilic peptide and protein-like drugs using lipidbased technologies[J].Ther Deliv,2011,2(12):1633-1653.
[8]Bernd Meibohm.Pharmacokinetic and pharmacodynamic evaluation of biologics:challenges and pitfalls[J].Chin J Clin Pharmacol Ther(中国临床药理学与治疗学),2007,12(10):1089-1098.
[9]Reddy ST,Berk DA,Jain RK,Swartz MA.A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles[J].J Appl Physiol,2006,101(4):1162-1169.
[10]Chen H,Langer R.Oral particulate delivery:status and future trends[J].Adv Drug Deliv Rev,1998,34(2-3):339-350.
[11]T Borchardt R,Jeffrey Aubé,Siahaan TJ,Gangwar S,Pauletti GM.Improvement of oral peptide bioavailability:peptidomimetics and prodrug strategies[J].Adv Drug Deliv Rev,1997,27(2-3):235-256.
[12]Rekha MR,Sharma CP.Oral delivery of therapeutic protein/peptide for diabetes-future perspectives[J].Int J Pharm,2013,440(1):48-62.
[13]Bernkop-Schnürch A,Schmitz T.Presystemic metabolism of orally administered peptide drugs and strategies to overcome it[J].Curr Drug Metab,2007,8(5):509-517.
[14]Maher S,Brayden DJ,Feighery L,McClean S.Cracking the junction:update on the progress of gastrointestinal absorption enhancement in the delivery of poorly absorbed drugs[J].Crit Rev Ther Drug Carrier Syst,2008,25(2):117-168.
[15]Kipnes M,Dandona P,Tripathy D,Still JG,Kosutic G.Control of postprandial plasma glucose by an oral insulin product(HIM2)in patients with type 2 diabetes[J].Diabetes Care,2003,26(2):421-426.
[16]Hui EJ,Yin BQ,Li ZF.Research development of oral peptide and protein drugs[J].Biotechnol Bull(生物技术通报),2011,6:21-24.
[17]Khafagy el-S,Morishita M,Ida N,Nishio R,Isowa K,Takayama K.Structural requirements of penetratin absorption enhancement efficiency for insulin delivery[J].J Control Release,2010,143(3):302-310.
[18]Khafagy el-S,Morishita M,Isowa K,Imai J,Takayama K.Effect of cell-penetrating peptides on the nasal absorption of insulin[J].J Control Release,2009,133(2):103-108.
[19]Guggi D,Kast CE,Bernkop-Schnürch A.In vivo evaluation of an oral salmon calcitonin-delivery system based on a thiolated chitosan carrier matrix[J].Pharm Res,2003,20(12):1989-1994.
[20]Wang XL,Tian H,Yao WB.Progress in the study of oral delivery of protein and peptide drugs[J].Pharm Biotechnol(药物生物技术),2011,18(5):445-448.
[21]Morishita M,Peppas NA.Is the oral route possible for peptide and protein drug delivery?[J].Drug Discov Today,2006,11(19-20):905-910.
[22]KV S, Devi GS, Mathew ST. Liposomal formulations of serratiopeptidase:in vitro studies using PAMPA and Caco-2 models[J].Mol Pharm,2008,5(1):92-97.
[23]Hanato J,Kuriyama K,Mizumoto T,Debari K,Hatanaka J,Onoue S,et al.Liposomal formulations of glucagon-like peptide-1:improved bioavailability and anti-diabetic effect[J].Int J Pharm,2009,382(1-2):111-116.
[24]Sarpietro MG,Castelli F.Transfer kinetics from colloidal drug carriers and liposomes to biomembrane models:DSC studies[J].J Pharm Bioallied Sci,2011,3(1):77-88.
[25]Zhang J,Zhang C,Gao S.The study of oral absorption and delivery systems for peptide drugs[J].Prog Pharm Sci(药学进展),2004,28(10):437-441.
[26]Cheng MB,Jin W,Wang JC,Chen DW,Zhang Q.Preparation and in vitro characterization of oral microemulsions containing earthworm fibrinolytic enzyme[J].Chin J New Drugs(中国新药杂志),2009,18(1):78-83.
[27]Kumar P, Mishra B. Colon targeted drug delivery systems-an overview[J].Curr Drug Deliv,2008,5(3):186-198.
[28]Page DT,Cudmore S.Innovations in oral gene delivery:challenges and potentials[J].Drug Discov Today,2001,6(2):92-101.
[29]Zhang W,Lv Z,Nie Z,Chen G,Chen J,Sheng Q,et al.Bioavailability of orally administered rhGM-CSF:a single-dose,randomized,open-label,two-period crossover trial[J].PLoS One,2009,4(5):e5353.
[30]Lan H,Nie Z,Liu Y,Lv Z,Liu Y,Quan Y,Chen J,et al.In vivo bioassay of recombinant human growth hormone synthesized in B.mori pupae[J].J Biomed Biotechnol,2010,2010:306462.
[31]Gong Z,Jin Y,Zhang Y.Suppression of diabetes in non-obese diabetic(NOD)mice by oral administration of a cholera toxin B subunit-insulin B chain fusion protein vaccine produced in silkworm[J].Vaccine,2007,25(8):1444-1451.
[32]Xiao HL,Zhang YZ,Wu XF.Oral administration activity determination of recombinant osteoprotegerin from silkworm larvae[J].Mol Biotechnol,2007,35(2):179-184.
[33]Liu T,Zhang YZ,Wu XF.High level expression of functionally active human lactoferrin in silkworm larvae[J].J Biotechnol,2005,118(3):246-256.
[34]Meng Q,Wang W,Shi X,Jin Y,Zhang Y.Protection against autoimmune diabetes by silkworm-produced GFP-tagged CTB-insulin fusion protein[J].Clin Dev Immunol,2011,2011:831704.
猜你喜欢
江苏安全生产(2022年8期)2022-11-01 09:14:32
昆明医科大学学报(2022年3期)2022-04-19 13:59:46
小资CHIC!ELEGANCE(2021年36期)2021-10-15 14:36:34
艺术评鉴(2020年5期)2020-04-30 06:47:57
人大建设(2018年10期)2018-12-07 01:13:46
中成药(2018年2期)2018-05-09 07:20:08
现代园艺(2017年13期)2018-01-19 02:28:09
中成药(2017年3期)2017-05-17 06:08:52
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
药学与临床研究(2015年4期)2015-06-05 11:35:54
