
麦冬多糖相对分子质量测定方法的研究
2013-11-01 03:18乔晚芳王月荣
中成药 2013年12期
胡 坪,乔晚芳,王 楠,陈 烨,黄 露,王月荣*
(1.上海市功能性材料化学重点实验室华东理工大学,上海 200237;2.华东理工大学化学与分子工程学院,上海 200237;3.马尔文仪器公司,上海 200237)
多糖的相对分子质量测定是其一级结构研究的重要内容,相对分子质量的大小显著影响多糖的性质及其生物活性。一般来说,多糖相对分子质量越大,分子体积越大,越不利于多糖跨越多重细胞膜障碍进入生物体内发挥生物学作用;但相对分子质量过低,又无法形成产生活性的聚合结构。因此,只有在一定的相对分子质量范围内,多糖才表现出其活性。不同多糖产生生物学活性的最佳相对分子质量范围不同[1]。
多糖相对分子质量及其分布的测定方法很多,如端基法、分析超离心法、黏度法、蒸气压渗透法、膜渗透压法、凝胶色谱、质谱法、光散射法等[1]。其中,端基法[2]要求试样中每一个多糖分子链端必须具有可以用化学方法分析定量的基团,且基团数目固定不变。分析超离心法[3]离心时间长,通常要几十小时。黏度法[4]是一种需要标准物质的相对测定方法,且测得的是多糖相对黏均分子质量。蒸气压渗透法[5]适合于相对分子质量小于3×104物质的相对分子质量测定,同时需要标准物质以测定K值。膜渗透压法对于相对分子质量较小的多糖测定误差较大。
高效凝胶色谱法(HPGPC)是一种大分子化合物的分离分析方法,具有设备和操作简单、分辨率高、重复性好等优点,已被药典收载用于测定多糖的相对分子质量与相对分子质量分布[6],是目前测定多糖相对分子质量及其分布最常用的方法。随着质谱分析仪器的不断发展,特别是基质辅助激光解吸/电离飞行时间质谱法(MALDI-TOF-MS)的出现,采用质谱法分析蛋白质及多糖等生物大分子的相对分子质量及其分布成为研究热点[7-8]。小角度激光光散射法是近年来发展起来的相对分子质量测定绝对方法。物质的相对分子质量直接由光散射信号通过Rayleigh光散射方程计算得到,因此不依赖于标准物质,无需做校正曲线。将分子排阻色谱与小角度激光光散射检测器法联用(SECLALLS),可以测得多糖混合样品的纯度和相对分子质量分布等,是一种理想的相对分子质量测定方法[9-10]。
麦冬多糖为中药麦冬Ophiopogon japonicus的主要成分之一。药理研究表明,麦冬多糖具有抗心肌缺血、降血糖、抗肿瘤、免疫活性、耐缺氧等多种药效[11-14]。本实验分别采用高效凝胶色谱联用蒸发光散射检测器法、基质辅助激光解吸/电离飞行时间质谱法和分子排阻色谱联用小角度激光光散射法,对麦冬多糖的相对分子质量及其分布进行测定和比较,为多糖化合物相对分子质量及其分布测定的方法选择提供参考,也可为麦冬多糖药物质量标准的制定提供依据。
1 仪器与试药
Agilent 1100高效凝胶色谱仪(Agilent,美国)配备 Alltech 3300 ELSD检测器(Grace,美国);4800 plus MALDI-TOF/TOF质谱仪(AB Sciex,美国);Viscotek GPCmax-TDA305多检测器(示差折光检测器、小角度激光光散射检测器、粘度检测器)GPC/SEC系统(Malvern,英国)。
浙麦冬Ophiopogon japonicus(购自浙江慈溪,由澳门科技大学姜志宏教授鉴定);DEAE-52(Whatman);Sephadex G-25、G-100(上海蓝季科技发展有限公司);标准葡聚糖T-1(MW=1000)、T-2(MW=5000)、T-3(MW=12000)、T-4(MW=50000)(Fluka,Switzerland);2,5-二羟基苯甲酸(Sigma,美国);硝酸钠(分析纯,国药集团化学试剂有限公司);聚氧乙烯、葡聚糖(Malvern,英国)。
2 方法与结果
2.1 麦冬粗多糖的提取和纯化[15]精密称取500 g浙麦冬药材,粉碎,过40目筛,以4倍量95%的乙醇回流提取1 h,冷却,抽滤,药渣置常温晾干。取50 g干燥药渣,加入500 mL去离子水,回流提取2 h,离心,上清液减压浓缩,加入95%乙醇,使提取液含醇量达80%,静置过夜,离心,沉淀冷冻干燥。取干燥沉淀适量,以去离子水溶解,依次用 DEAE-纤维素柱、Sephadex G100、Sephadex G25凝胶柱进行纯化,苯酚-硫酸法检测,绘制洗脱曲线,根据洗脱曲线合并峰位溶液,减压浓缩,冷冻干燥,得麦冬多糖。经苯酚-硫酸法测定,麦冬多糖的平均含有量为96.8% (n=3,RSD=0.7%)。
2.2 凝胶色谱测定方法 采用HPGPC-ELSD法对麦冬多糖的相对分子质量进行测定。方法为:取一定量的的麦冬多糖溶于纯净水中,配制成约1.0 mg/mL的溶液,用0.45μm水系滤膜过滤后,取续滤液注入凝胶色谱仪分析。色谱条件为:采用TSK-GEL G3000PW 凝胶色谱柱(10μm,7.5 mm×30 cm),流动相为纯水,体积流量 0.8 mL/min,进样量10μL,柱温30℃。ELSD气体流量为1.6 L/min,漂移管温度50℃,增益为1。结果麦冬多糖的凝胶色谱图(图1)中仅出现一对称狭窄峰,说明该多糖为均一多糖。
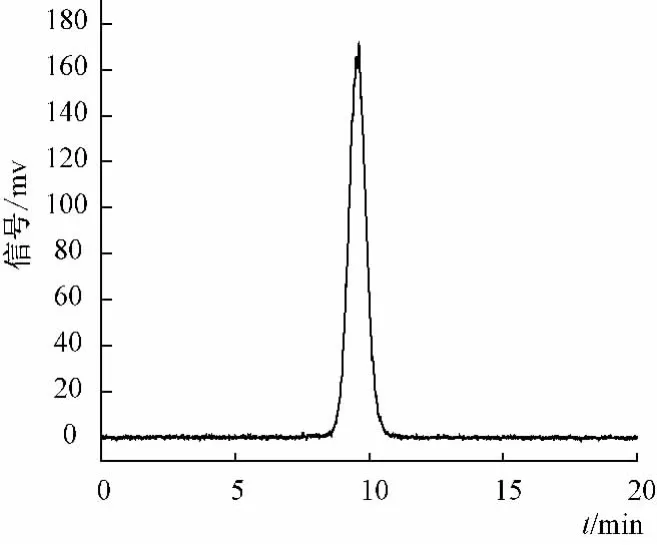
图1 麦冬多糖的HPGPC-ELSD谱图Fig.1 HPGPC-ELSD chromatogram of polysaccharides from Ophiopogon japonicus
分别称取一定量的标准葡聚糖T-1(Mw=1000)、T-2(Mw=5000)、T-3(Mw=12000)、T-4(Mw=50000),纯水溶解后,配成质量浓度约为1.0 mg/mL的溶液,取10μL注入凝胶色谱仪分析。以葡聚糖对照品的lgMw为纵坐标,以洗脱体积VR为横坐标,通过GPC软件绘制标准曲线,得到标准曲线方程为y=-0.623x+9.302(r2=0.990),计算得到麦冬多糖的Mn为1888 g/mol、Mw为 2376 g/mol、Mz为 2807 g/mol、Mp为 1971 g/mol,PD为1.23。分散系数 PD=Mw/Mn,用来表征分散程度。PD越大,说明相对分子质量越分散,PD=1,说明相对分子质量呈单分散。麦冬多糖的PD为1.23<2,表明其分散度较小。
2.3 MALDI-TOF-MS测定方法 采用基质辅助激光解吸/电离飞行时间质谱法对麦冬多糖的相对分子质量及其分布进行分析。方法为:将基质2,5-二羟基苯甲酸(DHB)及多糖样品均配成10 mg/mL水溶液,等体积混合均匀。采用滴液干燥法,取混合溶液0.5μL滴于样品靶上,室温晾干后,送入离子源进行测定。质谱条件为固体激光器频率200 Hz,反射正离子模式,谱图累加激光点400个。
图2为MALDI-TOF-MS结果的部分放大图。由图可见,每个聚合度的麦冬多糖出现3个系列的规律性加合离子峰,表1列出了图2中3个系列加合离子峰所对应的m/z值,每组系列峰中相邻两峰之间的m/z值均相差162,与1个己糖残基的质量相符,说明组成麦冬多糖的糖残基单元为己糖。S1系列可能为环状己聚糖或己聚糖脱水后的加钠离子峰,S2为线形己聚糖的加钠离子峰,S3为线形己聚糖的加钾离子峰。结果表明,麦冬多糖样品中主要存在线形和环状结构的己聚糖,相对分子质量分布范围主要在200~4000 g/mol之间。
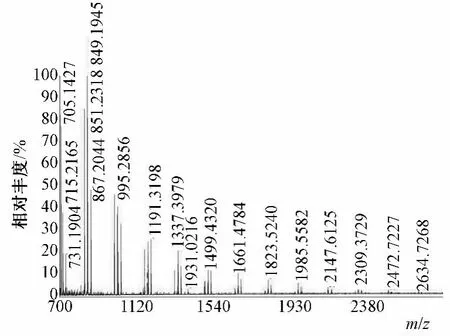
图2 麦冬多糖的MALDI-TOF-MS部分放大图Fig.2 Part enlarge MALDI-TOF-MS spectrum of polysaccharides from Ophiopogon japonicus
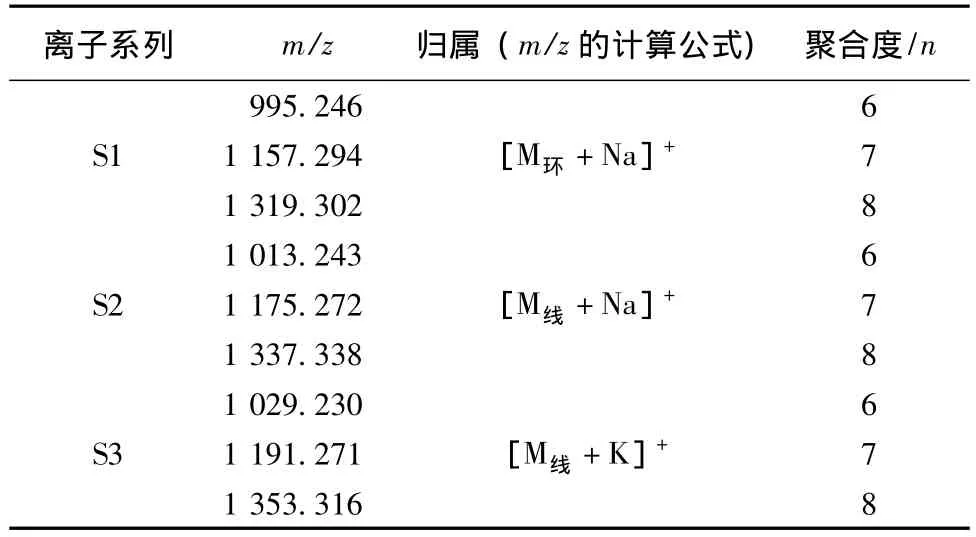
表1 麦冬多糖MALDI-TOF-MS图中3个系列离子峰归属Tab.1 Assignment of the ion peaks in MS spectrum of polysaccharides from Ophiopogon japonicus
通过MALDI-TOF-MS仪器的分析软件计算得到麦冬多糖的 Mn为486 g/mol、Mw为721 g/mol、Mz为1094 g/mol,分散度为1.49。
2.4 SEC-LALLS测定方法 采用Viscotek GPC-max-TDA305多检测器GPC/SEC系统对麦冬多糖的相对分子质量进行进一步分析。方法为:在室温条件下,将麦冬多糖(7.22 mg/mL)溶于0.1 mol/L NaNO3溶液中,经0.22μm微孔滤膜过滤后,取续滤液进GPC/SEC系统进行分析。采用1 x A6000M色谱柱(Viscotek),流动相为0.1 mol/L NaNO3,体积流量为0.7 mL/min,柱温为35℃,检测器温度为35℃,dn/dc值为0.14,对照品为聚氧乙烯和葡聚糖,进样量为100μL。
以SEC-LALLS法测定得到的麦冬多糖的三重检测器响应色谱图如图3所示。
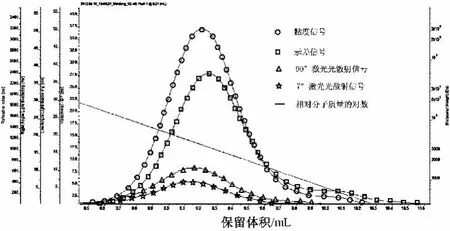
图3 麦冬多糖的SEC-LALLS色谱图Fig.3 SEC-LALLS spectrogram of polysaccharides from Ophiopogon japonicus
由图3可知,随着保留体积的增大,麦冬多糖相对分子质量的对数曲线从大到小分布,各个检测器的响应信号平滑、对称、没有拖尾,说明该SEC-LALLS分离、测定条件适合麦冬多糖的相对分子质量测定。由图3可见,黏度信号峰值最高点的保留体积小于示差检测器信号峰值最高点的保留体积,这是因为分子排阻色谱使得相对分子质量大的物质优先从色谱柱洗脱出来,同样的浓度下,对相对分子质量大小有响应的黏度信号大于示差检测器信号;随着麦冬多糖相对分子质量小的物质被洗脱出来,黏度信号小于示差检测器信号,符合各检测器响应特性规律。
麦冬多糖的重量百分比(相对分子质量分布)、累积重量比、Mark-Houwink曲线与相对分子质量(Da)的分析图见图4。麦冬多糖样品平行实验的计算结果如表2。
通过Rayleigh光散射方程和表2可得麦冬多糖的相对重均分子质量为3962 g/mol,平均分散度为1.49,流体力学半径为1.3 nm。图4中特性黏度曲线的斜率即为Mark-Houwink a值,a值小于0.3样品为球状结构,a值在0.7左右样品为无规卷曲结构,a值为0.45,说明麦冬多糖样品在水相的构象介于球状和无规卷曲之间。
3 讨论

图4 麦冬多糖的重量百分比(相对分子质量分布),累积重量比和Mark-Houwink曲线图谱Fig.4 Normalized weight fraction,Cumulativeweightfraction and Mark-Houwink spectrogram of polysaccharides from Ophiopogon japonicus
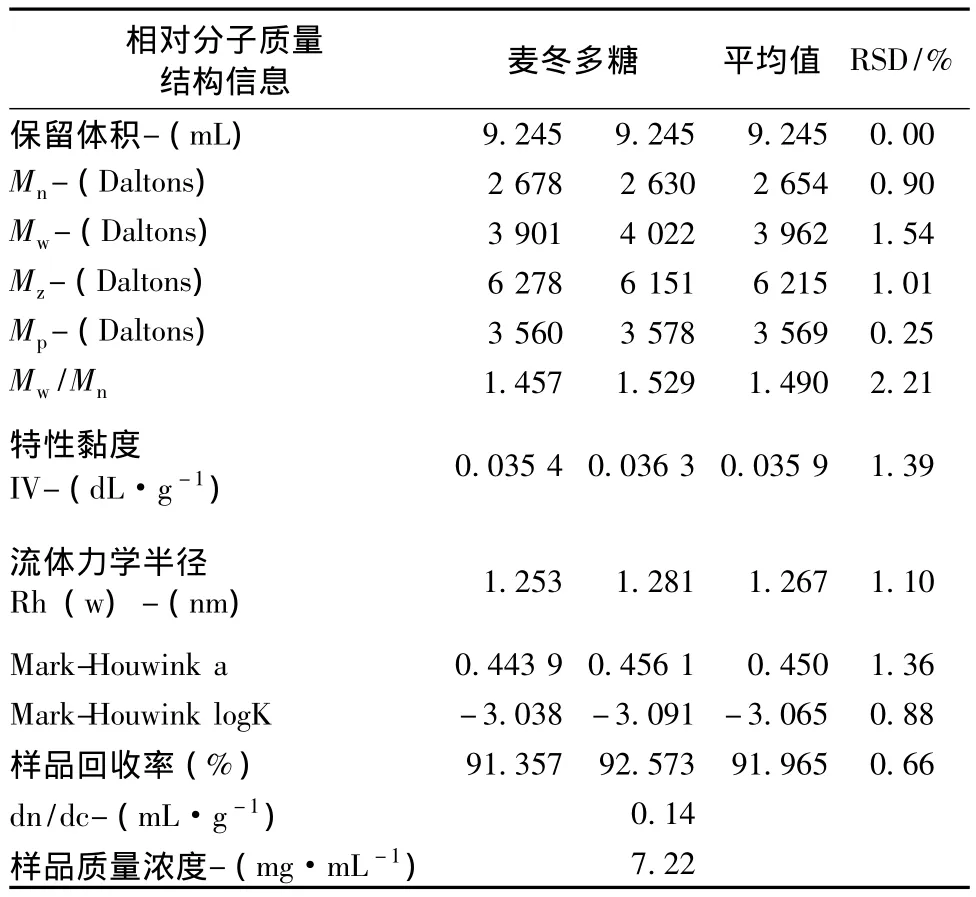
表2 麦冬多糖的SEC-LALLS测定结果Tab.2 SEC-LALLS determination results of polysaccharides from Ophiopogon japonicus
HPGPC-ELSD法是一种平均相对分子质量的相对测定方法。在测定过程中,需采用相对分子质量已知的多糖对照品建立LogMw-VR标准曲线,计算被测样品的相对分子质量。然而,凝胶色谱分离是基于尺寸筛分离原理,样品在色谱柱上的保留时间不仅与其相对分子质量有关,还与分子的空间结构相关。相对分子质量相同的线性结构和球状结构分子,表观尺寸差异很大,导致保留体积不同。可见,HPGPC-ELSD结果的准确性主要取决于样品和对照品在空间结构上的相似性。采用HPGPC-ELSD法测定的麦冬多糖相对分子质量结果较SECLALLS法小,是由于麦冬多糖样品在水相的构象介于球状和无规卷曲之间,与葡聚糖标样空间结构存在差异造成的。
MALDI-TOF-MS在检测模式上有正、负离子检测模式,多数情况下采用正离子检测模式,因为负离子模式下检测效率相对较低。检测方式有线性方式和反射方式两种,线性方式受低分子的影响较大,其主要测定相对分子质量大于10000 g/mol的物质,当相对分子质量小于10000 g/mol时,采用反射方式。麦冬多糖的相对分子质量分布在200~4000 g/mol之间,因此本试验在反射正离子模式下,对麦冬多糖相对分子质量及其分布进行了测定。MALDI-TOF-MS的相对分子质量测定结果与其他方法相比明显偏小,这可能是由于在低质荷比区域质谱图受基质干扰严重,且麦冬多糖的加合离子峰强度随m/z的增大而降低,使计算得到的平均相对分子质量偏低,分散系数增大。因此,MALDI-TOF-MS并不适合于测定相对分子质量较小的多糖的平均相对分子质量,但仍可以提供相对分子质量分布信息。同时,MALDI-TOF-MS还能够提供多糖的重复结构单元的相对分子质量信息,用于推测其结构、组成等。
SEC-LALLS可获得样品洗脱图中每一洗脱体积点的相对分子质量以及样品的相对分子质量分布。分子排阻色谱实现不同相对分子质量高聚物分子的分离,分子按大小顺序依次进入激光光散射检测器,通过Rayleigh光散射方程直接计算出相对分子质量Mw、Mn、Mz和 Mp及相对分子质量分布,不需要对照品对照以绘制标准曲线。该法操作简便,高效、误差小。而且黏度检测器还可以得到样品在溶液中的结构形态信息,包括流体力学体积Vh、流体力学半径 Rh、以及Mark-Houwink曲线、Mark-Houwink曲线a值、K值。本实验过程中,聚氧乙烯是窄相对分子质量分布标样,用来建立方法(包括测定仪器常数和校正各检测器之间的响应时间);葡聚糖是宽相对分子质量分布标样,用来验证聚氧乙烯建立的方法。dn/dc值(散射光角度和折光指数增量)与溶剂、样品单体结构、温度等因素有关,与样品相对分子质量无关,可通过阿贝折光仪测定得到。麦冬多糖在水相的dn/dc值通常处于0.13~0.15之间,因此本实验用0.14计算。
4 结论
本实验采用HPGPC-ELSD法、MALDI-TOF-MS法和SEC-LALLS法对麦冬多糖的相对分子质量及其分布进行了测定。HPGPC-ELSD法是一种相对分子质量相对测定法,多糖相对分子质量测定结果的准确性取决于样品与对照品结构的相似性。MALDI-TOF-MS法是一种相对分子质量测定的绝对方法,无需对照品,但测得的麦冬多糖相对重均分子质量误差较大,该方法更适合用于多糖单元结构等信息的获取。SEC-LALLS法也是一种相对分子质量测定的绝对方法,适合于多糖相对分子质量及分布测定,还可以获得样品在溶液中的结构形态信息,是一种较为理想的相对分子质量及其结构测定方法,但目前该仪器的普及率还不高。本实验的研究结果为多糖类化合物相对分子质量测定方法的选择提供了参考,也可为麦冬多糖药物质量标准的制定提供依据。
[1]戴 伟,刘新义,胡雄彬,等.香菇多糖的相对分子质量和结构与生物活性之间的关系[J].中南药学,2012,10(6):453-456.
[2]孟显丽,陈国华,侯 进,等.关于端基分析法测定壳低聚糖的相对数均分子质量问题的探讨[J].中国海洋大学学报,2005,35(1):142-144.
[3]倪寿亮.聚合物相对分子质量的测试与表征[J].广东化工,2012,39(2):190-193.
[4]何曼君,张红东,陈维孝,等.高分子物理[M].3版.上海:复旦大学出版社,2006:8-20.
[5]骆传环,黄荣清,刘曙晨.海星多糖的分子量测定[J].军事医学科学院院刊,2002,26(2):157-158.
[6]国家药典委员会.中华人民共和国药典:2010年版三部[S].北京:中国医药科技出版社,2010:附录20.
[7]Hu Wei,Fu Qiang,Zhou Jie,et al.Optimization of conditions for determination of protein molecular weight using matrixassisted laser desorption/ionization time-of-flight mass spectrometry[J].J South Agric,2012,43(1):14-17.
[8]Ham J S,Han G S,Jeong S G,et al.Determination of molecular weights of caprine milk proteins by matrix-assisted laser desorption/ionization mass spectrometry[J]. J Dairy Sci,2012,95(1):15-19.
[9]Krishnan G S,Burkanudeen A,Murali N,et al.Studies on molecular weight distribution of carbon fiber polymer precursors synthesized using mixed solvents[J].Chinese J Polym Sci,2012,30(5):664-673.
[10]韩凤梅,程伶俐,陈 勇.板桥党参多糖的分离纯化及组成研究[J].中国药学杂志,2005,40(18):1381-1383.
[11]Xiong Shuangli,Li Anlin,Huang Ni,et al.Antioxidant and immunoregulatory activity of different polysaccharide fractions from tuber of Ophiopogon japonicas[J].Carbohydr Polym,2011,86(3):1273-1280.
[12]Chen Xiaoming,Jin Jing,Tang Jia,et al.Extraction,purification,characterization and hypoglycemic activity of a polysaccharide isolated from the root of Ophiopogon japonicas[J].Carbohydr Polym,2011,83(2):749-754.
[13]Xu Jie, Wang Yuan, Xu Desheng, et al. Hypoglycemic effects of MDG-1,a polysaccharide derived from Ophiopogon ja-ponicas,in the ob/ob mouse model of type 2 diabetes mellitus[J].Int J Biol Macromol,2011,49(4):657-662.
[14]Zheng Qin,Feng Yi,Xu Desheng,et al.Influence of sulfation on anti-myocardial ischemic activity of Ophiopogon japonicuspolysaccharide[J].J Asian Nat Prod Res,2009,11(4):306-321.
[15]胡 坪,乔晚芳,沈骏文,等.麦冬多糖单糖组成的分析方法研究[J].药物分析杂志,2013,33(1):50-56.
猜你喜欢
四川蚕业(2022年2期)2022-11-19
玩具世界(2022年1期)2022-06-05
故事作文·高年级(2022年3期)2022-03-17
环境保护与循环经济(2021年7期)2021-11-02
火力与指挥控制(2018年10期)2018-11-13
中成药(2017年9期)2017-12-19
中国交通信息化(2017年9期)2017-06-06
中成药(2017年4期)2017-05-17
电子制作(2017年10期)2017-04-18
杂草学报(2015年2期)2016-01-04
