
二氢乳清酸脱氢酶靶向抗疟药研究进展
2013-10-27 09:04:26赵彩亮兰晶贝祝春杨恒林
生物技术通讯 2013年1期
赵彩亮 ,兰晶 ,贝祝春 ,杨恒林
1.大理学院 病原与媒介生物研究所,云南 大理 671003;2.军事医学科学院 微生物流行病研究所,北京 100071;3.云南省寄生虫病防治所,云南疟疾研究中心,云南 普洱 665000
疟疾是由疟原虫感染引起的虫媒传染病,是目前全球最受关注的公共卫生问题之一。全球每年约2.47亿人罹患疟疾,近100万人死于疟疾[1],婴儿、5岁以下儿童及孕妇是主要受威胁人群[2]。感染人类的疟原虫主要有间日疟原虫(Plasmodium vivax)、三日疟原虫(P.malariae)、恶性疟原虫(P.falciparum)和卵形疟原虫(P.ovale),其中以恶性疟原虫感染引起的恶性疟最为严重,是每年近百万人死于疟疾的罪魁祸首。
经过大量努力,疟疾在我国及欧美等发达国家已得到有效控制,但在非洲及东南亚部分国家仍广泛流行。20世纪60年代以来,恶性疟原虫对氯喹、磺胺多辛-乙胺嘧啶等广泛应用的抗疟药相继产生抗药性,全球疟疾的发病率和死亡率有所回升[3-4]。疟原虫抗药性的出现和快速传播,使得除青蒿素类药物之外的现有抗疟药普遍存在抗药性问题[5-6],且有研究表明已产生抗性的疟原虫株可更快地对新抗疟药产生抗药性[7]。为了应对日益严重的抗药性问题,遏制疟原虫抗药性的发展,世界卫生组织推荐以基于青蒿素类药物的复方疗法(artemisinin-based combination therapies,ACT)为无并发症性恶性疟的一线治疗方案,ACT已成为全球疟疾防控恶性疟的主要手段[8]。然而,随着某些地区恶性疟原虫对青蒿素类药物敏感性的日渐降低[9],使得这一目前惟一普遍有效的抗疟药也同样面临着疟原虫抗药性的危险。因此,寻找新的抗疟药,特别是与现有抗疟药靶点不同的新型抗疟药受到了越来越多的关注[2,5]。
近年来,有关学术和科研机构的支持推动了抗疟新药的研究[5,10-11],数种新的ACT已进入后期临床试验阶段,一些新抗疟药,如全合成的三恶烷类新药Rbx11160和OZ439、葛兰素史克研发的吡啶酮类新药GSK932121和默克授权MMV研发的MK4815也进入临床研究。恶性疟原虫基因组测序的完成,为寻找新的抗疟分子靶标带来了便利[12]。但已有研究所发现、鉴定的疟原虫必需或可药化(druggable)新靶点,却鲜有得到有效化合物的验证。阿托喹酮的作用靶点,线粒体电子转移链细胞色素bc1复合物的确证是临床有效抑制剂靶点发现的最近范例[13-16]。
许多临床使用的抗疟药通过直接或间接影响嘧啶代谢而发挥作用,如二氢叶酸还原酶或二氢叶酸合成酶抑制剂(如乙胺嘧啶、氯胍、磺胺类药物)阻断胸腺嘧啶合成所必需的叶酸的代谢[17]。阿托喹酮的作用靶点是线粒体电子转移链中的细胞色素bc1复合物,但阿托喹酮治疗导致细胞内UTP和CTP水平下降的发现[18-19],以及细胞色素bc1复合物活性对于为二氢乳清酸脱氢酶(dihydroorotate dehydroge⁃nase,DHODH)提供合成嘧啶所需氧化型辅酶Q的必需性[20-21],表明它同样通过阻断嘧啶代谢发挥抗疟作用。胸苷酸合成酶抑制剂虽未被用于临床,但也被证明有较强的抗疟作用[22-26]。上述研究表明,嘧啶生物合成途径是抗疟药发现的一个重要源泉。在此,我们简要综述近年来以恶性疟原虫嘧啶从头合成途径中第四个酶——DHODH为对象的抗疟新靶点及新药发现研究。
1 嘧啶从头合成途径为疟原虫所必需
作为DNA和RNA生物合成的前体,嘧啶是细胞生命所必需的代谢产物[21]。细胞可以氨、碳酸氢盐、左旋天冬氨酸为原料从头合成所需的嘧啶(从头合成途径),也可利用现成的嘧啶碱基或核苷酸(补救途径)。疟原虫较为特殊,它缺乏利用现成嘧啶的酶,从头合成途径是其获得细胞生长所需嘧啶的惟一来源。
疟原虫首先需要通过6个酶催化的生化反应合成UMP,后者进一步被用于ATP、CTP、cTMP及后续其他细胞生长所需的核苷酸代谢产物的合成。这6个酶分别为谷酰转氨-氨甲酰磷酸合成双功能酶(GAT/CPS)、天冬氨酸转氨甲酰酶(ACT)、二氢乳清酸酶(DHOtase)、二氢乳清酸脱氢酶(DHODH)、乳清酸磷酸核糖转移酶(OPRT)和乳清苷5'-单磷酸脱羧酶(OMPDC)。这些酶的存在已在疟原虫提取物酶活性的检测中得到证实[26-27],它们各自的编码基因也已在疟原虫基因组中得到鉴定[12]。其中,DHODH已从恶性疟原虫的线粒体中分离得到[28],而其基因序列在疟原虫基因组测序完成前就已被报道[29]。
疟原虫嘧啶补救途径的缺失最初是在核酸放射性标记前体掺入分析研究中发现的[30]。研究发现,乳清酸是惟一一种可被疟原虫利用的嘧啶合成前体。而后,5-氟乳清酸具有较强抗疟作用、5-氟尿嘧啶无抗疟作用的发现为此提供了佐证[23-24]。已完成的疟原虫基因组测序数据中无嘧啶补救途径相关基因的序列信息进一步证实这一点[12]。由于缺乏可利用现成嘧啶与核苷酸的补救途径,嘧啶从头合成途径中的所有酶都被认为是疟原虫所必需的。然而,要成为有价值的抗疟药靶点,除了应是疟原虫所必需的之外,还须满足以下几个条件:靶蛋白可以与类药小分子以高亲和力结合,即靶蛋白“可药化”;靶抑制剂能抑制细胞生长;抑制剂对疟原虫和宿主细胞有较高的选择性。如下所述,疟原虫DHODH可满足这些条件,因此被认为是理想的抗疟药新靶点。
2 恶性疟原虫DHODH是理想的抗疟药新靶点
DHODH属于一类带有β/α桶状结构域的蛋白质超家族[31]。不同物种中的DHODH有Ⅰ型(位于细胞浆,如细菌)和Ⅱ型(位于线粒体内膜或细胞膜)2种存在类型。恶性疟原虫DHODH(PfDHODH)和人DHODH同为Ⅱ型线粒体酶,人DHODH作为药物靶点的有效性已得到临床验证,以此为靶点的莱氟米特(Leflunomide)已获准用于治疗类风湿关节炎[32],布奎那(Brequinar)(图1)也作为抗肿瘤药进入了临床研究[33]。这为同为线粒体型酶的PfDHODH作为药物靶点的可行性提供了“可药化”数据支持。
在嘧啶从头合成中,DHODH以黄素单核苷酸(flavin mononucleotide,FMN)依赖的氧化-还原反应催化二氢乳清酸形成乳清酸,这需要2步催化反应来完成:①FMN还原驱动的二氢乳清酸氧化;②通过还原型黄素单核苷酸(FMNH2)的再氧化使酶重新活化。FMN氧化-还原循环所需的辅助因子因酶类型和细胞定位的不同而有所不同。Ⅰ型胞浆酶须利用富马酸或NAD+,Ⅱ型线粒体酶则通过利用氧化型辅酶Q而与线粒体电子转移链偶合[20]。最近,Vaidya等[21-22]验证了疟原虫嘧啶从头合成与线粒体电子转移链间的这种密切联系。通过重组表达富马酸依赖的胞浆型酵母DHODH,恶性疟原虫可绕过对自身线粒体型酶的依赖,从而表现出阿托喹酮抗性。这表明至少就疟原虫的体外生长而言,细胞色素bc1复合物的主要功能是为DHODH催化产生乳清酸提供氧化型辅酶Q,从而证明了DHODH是疟原虫所必需的,且通过干扰DHODH催化的生化反应可抑制疟原虫的生长,如阿托喹酮。
一些对人DHODH有较强抑制作用的抑制剂对PfDHODH并无抑制活性,这为DHODH抑制剂的选择性作用提供了线索[34]。随后的X线衍射结构分析表明,虽然A77 1726(莱氟米特的活性代谢物,图1)与PfDHODH和人DHODH结合于相同的口袋状结构,但二者与抑制剂结合的氨基酸残基并不具有保守性[35]。因此,寻找具有选择性作用的DHODH抑制剂是切实可行的。此外,嘧啶补救途径的缺失使疟原虫完全依赖于DHODH参与的嘧啶从头合成途径,进而对DHODH抑制剂敏感,而人类细胞却可通过嘧啶补救途径获取维持细胞生长所需的嘧啶。这也使DHODH抑制剂具有较高的选择性。
正如上所述,PfDHODH作为理想的抗疟药发现新靶点受到了越来越多的关注。高通量筛选(high-throughput screening,HTS)、已 知 DHODH 抑制剂的药物化学研究和基于酶-配体复合物的晶体结构解析研究等相继被用于PfDHODH选择性抑制剂的筛选、鉴定。其中,HTS取得了较大的成功。
3 PfDHODH抑制剂的高通量筛选研究
2005年,美国德州大学西南医学中心Phillips领导的研究小组首次报道了PfDHODH抑制剂HTS研究[34]。他们建立了基于二氯靛酚(dichloroindophe⁃nol,DCIP)还原染色法的PfDHODH抑制剂HTS方法,并对220 000个化合物进行了筛选。以3 μmol/L的单浓度样品进行筛选,获得了1249个命中化合物(抑制率>60%),命中率达0.6%。相同浓度的人DHODH抑制活性测定表明,几乎所有的命中物都表现出对PfDHODH的选择性抑制作用。初步的量效关系测定表明,其中IC50<0.6 μmol/L的包括苯基苯甲酰胺类(phenylbenzamides)、尿素类(ureas)、萘甲酰胺类化合物(naphthamides)和三唑并嘧啶(trizolo⁃pyrimidine)系列化合物(图2,a~d)。其中,前3类是命中物中最常见的化合物类型,其对PfDHODH的IC50为0.02~0.8 μmol/L,且比对人DHODH的抑制活性高70~12 500倍,但对体外培养的恶性疟原虫并无抑制作用,因此未得到进一步的研究。之后,哈佛大学的Clardy等对含208 000个结构样本的小分子化合物进行了PfDHODH抑制活性的HTS研究[36]。同样用基于DCIP还原染色的方法,以10 μmol/L的单浓度样本进行筛选,命中率达0.3%,其中55个命中化合物IC50在1 μmol/L以下,5类化合物结构在体外培养恶性疟原虫试验中表现出较强的抗疟作用,并具有良好的作用选择性。其中活性最强的为噻吩类化合物(图2,e;PfDHODH IC50=40 nmol/L),但进一步的系列化合物合成与初步构效关系研究并未发现活性更强的类似物。
4 基于人DHODH抑制剂的药物化学研究
英国利兹大学的一个研究小组对一系列布奎那的衍生物进行了探索研究。布奎那对PfDHODH的活性极低(IC50>500 μmol/L),其衍生物中活性最强的也只达到中等活性(IC50=40 μmol/L),且作用选择性不高[37]。他们以A77 1726结构骨架为模型,设计、合成高效、选择性PfDHODH抑制剂的尝试也同样未获得成功[38-40]。最终,他们以四环胺为起点获得了一系列活性达亚微摩尔级别(IC50=0.2~0.4 μmol/L)且对PfDHODH具有较高选择性(200~1200倍)的衍生物。其中活性最强的体外抗恶性疟原虫3D7的EC50为 2 μmol/L(图2,f)。
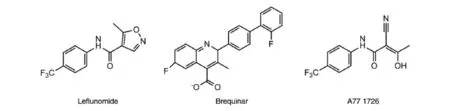
图1 人DHODH抑制剂
5 三唑并嘧啶类PfDHODH抑制剂的研究
Phillips等[36]发现三唑并嘧啶类化合物DSM1(图2,d)具有较高的作用选择性,且在体外培养恶性疟原虫试验中同样具有较强活性(IC50PfDHODH=0.047 μmol/L;IC50人 DHODH>200 μmol/L;EC50恶性疟原虫3D7=0.079 μmol/L)。在此基础上,他们建立了一个简单、廉价的三步合成路线,合成若干DSM1类似物,并开展了进一步的研究。他们的研究为PfDHODH抑制剂的抗疟研究及PfDHODH靶向抗疟新药的深入研究提供了大量有用的信息。
5.1 构效关系研究
结构优化和构效关系(SAR)研究数据表明[41-43]:①无取代的桥氮N1是化合物活性所必需的;②在萘基环引入杂原子导致化合物活性降低;③可用大芳香环或疏水环系统,如蒽、取代苯环等替化合物结构中的萘基;④替代萘基的取代苯环其取代位置只能是对位(CF3>Br>Cl>Me>F),邻位取代导致活性的完成丧失;⑤C6位的乙基或三氟甲基取代和C2位的甲基取代导致化合物活性降低。而且,这一系列化合物的酶抑制活性与其体外抗疟活性存在较好的相关性。这说明DHODH是它们发挥抗疟作用的细胞靶点。
5.2 X线衍射晶体结构研究
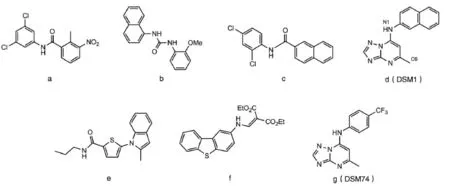
图2 PfDHODH抑制剂[IC50(μmol/L),PfDHODH vs人DHODH]

图3 PfDHODH及人DHODH的X线衍射晶体结构
PfDHODH与三唑并嘧啶类系列化合物结合复合物的X线衍射结构解析研究提供了小分子与PfDHODH选择性结合的结构信息[42]。首先,PfDHODH与三唑并嘧啶系列抑制剂的结合位点由2部分组成(图3),一个与萘基、蒽基或三氟甲基-苯基结合的完全疏水位点和能与三唑并嘧啶形成氢键相互作用的口袋状结构。萘基与2个苯丙氨酸残基(Phe227和Phe188)通过π键之间的堆积力相互作用,桥氮N1和三唑并嘧啶环分别与His185和Arg265形成氢键相互作用。其中,三唑并嘧啶基团的位置与A77 1726与PfDHODH结合的β-羟基烯胺部分重叠[35]。但萘基的结合位点是通过Phe188的旋转形成的一个新的口袋状结构。这解释了为何PfDHODH能与多种结构类型化合物以高亲和力结合。其次,三唑并嘧啶类化合物与PfDHODH结合的某些关键氨基酸残基在不同物种间并不具保守性,与人DHODH结合抑制剂的残基位点也不相同(图3)。如在接近疏水口袋处,A77 1726的三氟甲基苯基结合于人DHODH的Ala59、Gly363和Pro364,而三唑并嘧啶类化合物的萘基与PfDHODH的Phe188和Met536形成相互作用,且因Phe188的旋转与Leu189和Leu197形成一个新的萘基结合口袋。这为三唑并嘧啶类化合物对PfDHODH的选择性结合提供了结构基础。此外,配体结合DHODH与小分子的晶体结构比较显示桥氮N1的强斥电子作用使三唑并嘧啶环电荷分布偏移而更利于与相应氨基酸残基发生稳定的氢键相互作用。以不能形成上述分子内电荷偏移的O或S取代N1使化合物活性丧失,及His185或Arg265突变为Ala后DHODH与抑制剂的结合力降低,说明这种稳定氢键相互作用对于三唑并嘧啶类化合物的活性至关重要。最后,从三唑并嘧啶环至FMN结合位置之间存在一个狭窄的沟状结构空间,提示先导化合物的进一步结构优化也许可利用这一结构空间的特征。
5.3 体内抗疟作用的研究
以标准鼠疟模型进行的体内抗疟研究发现,原始的HTS命中化合物DSM1虽然在细胞试验中表现出了较强的抗疟作用,对伯氏疟原虫(P.berghei)感染却无抑制作用。药代动力学研究发现多次给药可导致血药浓度的显著下降,提示DSM1可能是一个代谢诱导物。这被认为是其在动物模型中无效的原因。此外,部分三唑并嘧啶系列化合物对恶性疟原虫和伯氏疟原虫DHODH抑制活性的差异也被认为是其在动物模型中无效的可能原因。基于此,Phil⁃lips等[43]进行了进一步的交互式先导化合物优化研究,以不同的取代苯环替换萘基,合成系列类似物,并用人肝脏微粒体模型测试其代谢稳定性。最终,他们获得了一个代谢稳定,且在鼠疟模型中具有显著抗疟作用的三唑并嘧啶类化合物DSM74(图2:g)。在以伯氏疟原虫感染小鼠模型进行的标准4 d抑制试验中,DSM74 50 mg/kg每天2次和每天1次的抑制率分别达到了95%和71%,且具有良好的耐受性无明显的毒副作用。
这些数据表明PfDHODH抑制剂在体内可有效发挥作用并抑制疟原虫血症,进一步确证了PfDHODH作为抗疟新药发现靶点的有效性。
6 结语
嘧啶补救途径的缺失使疟原虫对嘧啶从头合成途径靶向抑制剂生物高度敏感。DHODH催化嘧啶从头合成途径的第四步反应,其作为疟原虫必需的酶和可药化靶点已得到了遗传学和化学研究的证实。PfDHODH作为抗疟药发现的新靶点受到了越来越多的关注,其中HTS作为PfDHODH抑制剂筛选、鉴定的有效策略已取得了很大的成功。X线衍射晶体结构解析的研究为PfDHODH选择性抑制剂的鉴定、设计提供了结构学基础,并为先导化合物的进一步优化提供了大量有价值的信息。三唑并嘧啶系列化合物已经从命中化合物阶段进入了早期先导物优化研究阶段,并已获得了具有良好代谢稳定性和体内抗疟活性的类似物。综上所述,PfDHODH是一个有前景的抗疟新药研究靶点,PfDHODH抑制剂研究的成功将为临床提供与现有抗疟药全然不同的新型抗疟药。
[1]World Health Organization.World Malaria Report 2008[M].Switzerland:World Health Organization,2008.
[2]Burrows J N,Chibale K,Wells T N.The state of the art in anti-malarialdrug discovery and development[J].CurrTop Med Chem,2011,11:1226-1254.
[3]杨恒林,刘德全,黄开国,等.云南省恶性疟原虫对氯喹、氨酚喹、哌喹、甲氟喹、奎宁敏感性的体外测定[J].中国寄生虫学与寄生虫病杂志,1999,17:43-45.
[4]Greenwood B M,Fidock D A,Kyle D E,et al.Malaria:prog⁃ress,perils,and prospects for eradication[J].J Clin Invest,2008,118(4):1266-1276.
[5]Craft J C.Challenges facing drug development for malaria[J].Curr Opin Microbiol,2008,11(5):428-433.
[6]White N J.Antimalarialdrug resistance[J].JClin Invest,2004,113(8):1084-1092.
[7]Rathod P K,McErlean T,Lee P C.Variations in frequencies ofdrug resistance in Plasmodium falciparum[J].Proc Natl Acad Sci USA,1997,94(7):9389-9393.
[8]White N J.The role of anti-malarial drugs in eliminating ma⁃laria[J].Malar J,2008,11(7):S8.
[9]Yang H L,Liu D Q,Yang Y M,et al.Changes in suscepti⁃bility of Plasmodium falciparum to artesunate in vitro Yunnan province,China[J].Trans Roy Soc Trop Med Hyg,2003,97:226-229.
[10]Gelb M H.Drug discovery for malaria:a very challenging and timely endeavor[J].CurrOpin Chem Biol,2007,11(4):440-445.
[11]Olliaro P,Wells T N.The global portfolio of new antimalarial medicines under development[J].Clin Pharmacol Ther,2009,85(6):584-595.
[12]Gardner M J,Hall N,Fung E,et al.Genome sequence of the human malaria parasite Plasmodium falciparum[J].Nature,2002,419(6906):498-511.
[13]FryM,Pudney M.Site of action of the antimalarial hy⁃droxynaphthoquinone, 2-[trans-4-(4'-chlorophenyl)cyclohex⁃yl]-3-hydroxy-1,4-naphthoquinone(566C80)[J].Biochem Phar⁃macol,1992,43(7):1545-1553.
[14]Mather M W,Darrouzet E,Valkova-Valchanova M,et al.Un⁃covering the molecularmode ofaction ofthe antimalarial drugatovaquoneusinga bacterialsystem[J].JBiolChem,2005,280(29):27458-27465.
[15]Srivastava I,Morrisey J,Darrouzet E,et al.Resistance muta⁃tions reveal the atovaquone-binding domain of cytochrome b in malaria parasites[J].Mol Microb,1999,33(4):704-711.
[16]Srivastava I,Rottenberg H,Vaidya A.Atovaquone,a broad spectrum antiparasitic drug,collapses mitochondrial membrane potential in a malarial parasite[J].J Biol Chem,1997,272(7):3961-3966.
[17]Hyde J E.Targeting purine and pyrimidine metabolism in hu⁃man apicomplexan parasites[J].Curr Drug Targets,2007,8(1):31-47.
[18]Seymour K K,Yeo A E,Rieckmann K H,et al.dCTP levels are maintained in Plasmodium falciparum subjected to pyrimi⁃dine deficiency or excess[J].Ann Trop Med Parasitol,1997,91(6):603-609.
[19]Ittarat I,Asawamahasakda W,Meshnick S R.The effects of antimalarials on the Plasmodium falciparum dihydroorotate de⁃hydrogenase[J].Exp Parasitol,1994,79(1):50-56.
[20]Hyde J E.Targeting purine and pyrimidine metabolism in hu⁃man apicomplexan parasites[J].Curr Drug Targets,2007,8(1):31-47.
[21]Painter H J,Morrisey J M,Mather M W,et al.Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum[J].Nature,2007,446(7131):88-91.
[22]Vaidya A B,Mather M W.Mitochondrial evolution and func⁃tions in malaria parasites[J].Annu Rev Microbiol,2009,63:249-267.
[23]Rathod P K,Khatri A,Hubbert T,et al.Selective activity of 5-fluoroorotic acid against Plasmodium falciparum in vitro[J].Antimicrob Agents Chemother,1989,33(7):1090-1094.
[24]Rathod P K,Leffers N P,Young R D.Molecular targets of 5-fluoroorotate in the human malaria parasite,Plasmodium fal⁃ciparum[J].Antimicrob Agents Chemother,1992,36(4):704-711.
[25]Rathod P K,Reshmi S.Suceptibility of Plasmodium falci⁃parum to a combination of thymidine and ICI D1694,a quin⁃azoline antifolate directed at thymidylate synthase[J].Antimi⁃crob Agents Chemother,1994,38(3):476-480.
[26]Rathod P K,Reyes P.Orotidylate-metabolizing enzymes of thehuman malarialparasite,Plasmodium falciparum,differ from host cell enzymes[J].J Biol Chem,1983,258(5):2852-2855.
[27]Reyes P,Rathod P K,Sanchez D J,et al.Enzymes of pu⁃rine and pyrimidine metabolism from the human malaria para⁃site,Plasmodium falciparum[J].Mol Biochem Parasitol,1982;5(5):275-290.
[28]Krungkrai J.Purification,characterization and localization of mitochondrial dihydroorotate dehydrogenase in Plasmodium fal⁃ciparum,human malarial parasite[J].Biochim Biophys Acta,1995,1243(3):351-360.
[29]LeBlanc S B,Wilson C M.The dihydroorotate dehydrogenase genehomologueofPlasmodium falciparum[J].MolBiochem Parasitol,1993,60(2):349-352.
[30]Gutteridge W E,Trigg P I.Incorporatin of radioactive precur⁃sors into DNA and RNA of Plasmodium knowlesi in vitro[J].J Protozool,1970,17(1):89-96.
[31]Nara T,Hshimoto T,Aoki T.Evolutionary implications of the mosaic pyrimidine-biosynthetic pathway in eukaryotes[J].Gene,2000,257(2):209-222.
[32]Herrmann M L,Schleyerbach R,Kirschbaum B J.Lefluno⁃mide:an immunomodulatory drug for the treatment of rheuma⁃toid arthritis and other autoimmune diseases[J]. Immuno⁃pharm,2000,47(2-3):273-289.
[33]Peters G J,Schwartsmann G,Nadal J C,et al.In vivo inhibi⁃tion of the pyrimidine de novo enzyme dihydroorotic acid de⁃hydrogenase by brequinar sodium(DUP-785;NSC 368390)in mice and patients[J].Cancer Res,1990,50(15):4644-4649.
[34]Baldwin J,Farajallah A M,Malmquist N A,et al.Malarial di⁃hydroorotate dehydrogenase:substrate and inhibitor specificity[J].J Biol Chem,2002,277(44):41827-41834.
[35]Hurt D E,Widom J,Clardy J.Structure of Plasmodium falci⁃parum dihydroorotate dehydrogenase with a bound inhibitor[J].Acta Crystallogr D Biol Crystallogr,2006,62(Pt 3):312-323.
[36]Patel V,Booker M,Kramer M,et al.Identification and char⁃acterization of small molecule inhibitors of Plasmodium falci⁃parum dihydroorotate dehydrogenase[J].J Biol Chem,2008,283(50):35078-35085.
[37]Boa A N,Canavan S P,Hirst P R,et al.Synthesis of brequi⁃nar analogue inhibitors of malaria parasite dihydroorotate dehy⁃drogenase[J].Bioorg Med Chem,2005,13(6):1945-1967.
[38]Heikkilae T,Thirumalairajan S,Davies M,et al.The first de novo designed inhibitors of Plasmodium falciparum dihydrooro⁃tate dehydrogenase[J].Bioorg Med Chem Lett,2006,16(1):88-92.
[39]Heikkila T,Ramsey C,Davies M,et al.Design and synthesis of potent inhibitors of the malaria parasite dihydroorotate dehy⁃drogenase[J].J Med Chem,2007,50(2):186-191.
[40]Davies M,Heikkila T,McConkey G A,et al.Structure-based design,synthesis,and characterization of inhibitors of human and Plasmodium falciparum dihydroorotate dehydrogenases[J].J Med Chem,2009,52(9):2683-2693.
[41]Phillips M A,Gujjar R,Malmquist N A,et al.Triazolopyrimi⁃dine-based dihydroorotate dehydrogenase inhibitorswith po⁃tent and selective activity against the malaria parasite,Plasmo⁃dium falciparum[J].J Med Chem,2008,51(12):3649-3653.
[42]Deng X,Gujjar R,El Mazouni F,et al.Structural plasticity of malaria dihydroorotate dehydrogenase allows selective bind⁃ing of diverse chemical scaffolds[J].J Biol Chem,2009,284(39):26999-27009.
[43]Gujjar R,Marwaha A,El Mazouni F,et al.Identification of a metabolically stabletriazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice[J].J Med Chem,2009,52(7):1864-1872.
猜你喜欢
健康体检与管理(2022年2期)2022-04-15 23:57:24
发明与创新(2020年5期)2020-12-20 23:31:23
科学导报(2020年69期)2020-11-09 03:38:44
天然产物研究与开发(2019年1期)2019-03-01 05:41:10
郑州大学学报(医学版)(2018年5期)2018-10-10 10:45:20
郑州大学学报(医学版)(2018年5期)2018-10-10 10:45:20
中国感染与化疗杂志(2018年6期)2018-01-19 23:23:10
汉语世界(2017年5期)2017-09-21 07:44:38
永善文学(2016年4期)2016-11-19 09:41:08
中国医学创新(2014年9期)2014-04-24 11:13:32
