
米曲霉(Aspergillus oryzae)RIB40中烯酮/烯酯还原酶的异源表达及性质分析
2013-10-25 06:25张海灵高秀珍冯进辉张同存吴洽庆朱敦明
生物加工过程 2013年1期
张海灵,高秀珍,陈 曦,任 杰,冯进辉,张同存,吴洽庆,朱敦明
(1.天津科技大学 生物工程学院,天津 300457;2.中国科学院 天津工业生物技术研究所 工业酶国家工程实验室,天津 300308)
烯烃的不对称加氢反应可以同时引入两个手性中心,在不对称合成技术中是非常重要的一类反应,是合成很多精细化工品的必要步骤。目前以金属催化剂为媒介的金属催化不对称加氢反应应用较为广泛,但是金属催化过程所需要的金属催化剂价格昂贵,氢气易燃易爆,产物中残留重金属,产品构型受限制等限制了其应用[1-2],烯酮/烯酯还原酶则是一类可以温和实现C=C不对称还原的生物催化剂,可以与金属催化互补[3]。目前利用烯酮/烯酯还原酶催化的有价值的反应大都是用全细胞来催化完成的[4-7]。利用微生物细胞虽然可以高立体选择性地还原多种α,β-不饱和醛或酮,但是对还原C=C键和C=O键的化学选择性通常较弱,这是因为细胞中还存在醇脱氢酶,从而产生副产物,导致总产率降低[8-9]。为了排除这一干扰,近几年来人们倾向于对烯酮/烯酯还原酶进行分离纯化后从事底物谱及其应用研究,从而最大限度地消除各种副反应的发生,这不仅增强反应的化学和立体选择性,而且还大大增加了产率[10-13]。
笔者对Aspergillus oryzae RIB40中的烯酮/烯酯还原酶进行了异源表达,镍柱纯化的蛋白进行酶学性质及底物谱的分析。
1 材料与方法
1.1 材料和仪器
1.1.1 菌株和质粒
米曲霉A.oryzae RIB40,由华南理工大学潘力教授提供;宿主菌大肠杆菌 E.coli DH5α、E.coli BL21(DE3)和克隆载体pUC19,由中国科学院工业酶国家工程实验室保存;表达载体pET32a(+),购自Novagen公司。
1.1.2 培养基
YPD培养基 (g/L):酵母提取物5,蛋白胨10,葡萄糖 20,KCl 2,KH2PO41,MgSO4·7H2O 0.5,FeSO4·7H2O 0.02;pH 5.5。
LB培养基(g/L):酵母提取物5,蛋白胨10,NaCl 10;pH 7.0。
抗生素使用质量浓度为100 μg/mL,使用氨苄青霉素(Amp)。
1.1.3 主要试剂和仪器
限制性内切酶 KpnⅠ、EcoRⅠ、SmaⅠ,购自Fermentas公司;PrimeSTAR HS DNA聚合酶和DNA连接试剂盒,购自TaKaRa公司;RNA酶,购自Sigma公司;DNA凝胶回收试剂盒,购自天根生化科技(北京)有限公司;NAD(P)H和 NAD(P),购自美国Codexis公司;所有的底物,购自Alfa Aesar或Sigma Aldrich公司;核酸染色剂GelRed,购自Biotium Inc公司;质粒提取试剂盒和蛋白定量试剂盒,购自北京康为试剂有限公司。
离心机分别购自美国Eppendorf和Thermo公司;高压匀浆机,购自德国APV公司;蛋白纯化仪和Superdex 20010/300 GL,购自美国GE公司;酶标仪SPECTRAMAX M2e,购自美国MD公司;气相色谱仪Agilent 7890,购自安捷伦公司;手性柱CP ChiraSil DEX(25 m ×0.25 mm ×0.25 μm),购自德国Varian 公司。
1.2 方法
1.2.1 基因组DNA的提取
A.oryzae RIB40菌株经YPD液体培养基培养2 d(30℃、200 r/min)后,4000 r/min 4℃离心10 min收集菌体;菌体冻干后液氮研磨成粉末,溶于600 μL 裂解液(50 mmol/L NaOH,1 mmol/L EDTA,1%Triton X-100)中重悬,并加入体积分数1%β-巯基乙醇;65℃孵育1 h后,12000 r/min离心10 min;上清用酚 -氯仿萃取2次后,异丙醇沉淀DNA,所得DNA沉淀用含有100 μg/mL RNA酶的重蒸水重悬,37℃孵育1 h以便DNA能完全溶解。
1.2.2 基因asper的克隆
根据 A.oryzae RIB40烯酮/烯酯还原酶基因(Genbank ID:XM_001727598.1)序列设计引物为:5'-GGGGTACCGACGACGACGACAAGGGATCCATCTCTTCCACATC-3'(下划线部分为KpnⅠ酶切位点,黑体部分为肠激酶酶切位点)和5'-CGGAATTCCTAAAGTGCACGCCAGAACT-3'(下划线部分为EcoRⅠ酶切位点)。以A.oryzae RIB40基因组DNA为模板,PCR扩增条件为:94℃预变性5 min;98℃,30 s;55℃,45 s;72℃,100 s;30个循环。扩增产物用含有核酸染色剂GelRed的0.8%琼脂糖凝胶电泳检测并回收。纯化后的DNA片段连接到pUC19(SmaⅠ单酶切)线性载体上,连接产物转入E.coli DH5α感受态细胞中,在含有100 mg/L氨苄青霉素、40 mg/L X-gal和100 μmol/L IPTG的LB平板上37℃培养16 h,随机挑选白色克隆进行PCR验证。阳性克隆质粒进行测序和序列分析,命名为pUC-asper。
1.2.3 asper基因表达载体的构建与蛋白AspER的表达纯化
重组质粒pUC-asper经限制性内切酶KpnⅠ和EcoRⅠ消化后连接到同样内切酶消化的pET32a(+)上,得到的重组质粒pET32a(+)-asper转入 E.coli DH5α感受态细胞中,在含有Amp抗性的LB平板上37℃培养,提质粒后进行双酶切验证,将阳性质粒转化到E.coli BL21(DE3)中得到重组工程菌。
将单菌落接种于含有Amp的LB培养液的试管中,37℃培养过夜;以1%接种量接种于含有同样抗生素的800 mL LB培养基,37℃下200 r/min震荡培养至OD600为0.6~0.8时加入0.1 mmol/L IPTG,37℃诱导表达6 h。培养后的菌体在7000 r/min离心15 min的条件收集后洗涤并重悬于缓冲液A(20 mmol/L Tris-HCl pH 7.4,250 mmol/L NaCl)中,高压匀浆破碎细胞。所得细胞裂解液在4℃下经14000 r/min离心30 min后取上清,0.45 μm滤膜过滤后进行亲和层析纯化目的蛋白,咪唑浓度梯度洗脱获得目的蛋白。所得目的蛋白进行SDS-PAGE检测,并根据蛋白定量试剂盒的操作流程测定蛋白浓度,随后加入50%甘油保存在-20℃中备用。
1.2.4 酶活测定及动力学参数测定
AspER酶活性的标准检测采用微孔板反应,在酶标仪上进行。反应体系总体积为200 μL,包含5 mmol/L 2-环己烯酮(DMSO溶解),0.5 mmol/L NADPH和100 μg/mL AspER。反应体系的配制采用20 mmol/L Tris-HCl缓冲液(pH 7.4)。反应的发生以加入NADPH作为启动。反应通过连续监测25℃下340 nm吸光值的变化来测定酶对底物的活性大小。NADPH摩尔消光系数为6.22/(mmol·cm)。一个酶活力单位(U)定义为每分钟内消耗1 μmol NADPH所需的酶量。
AspER对底物2-环己烯酮的动力学参数测定方法同样按照上述酶活分析方法进行。2-环己烯酮的浓度范围设定为0.1~10 mmol/L。反应速度定义为每分钟消耗NADPH的毫摩尔数。采用双倒数法计算Km和Vmax。
1.2.5 酶聚集状态的确定
AspER在天然状态下的聚集状态通过结合分子排阻来进行确定。所用层析柱为Superdex 20010/300 GL,流动相为50 mmol/L磷酸盐缓冲液(含有150 mmol/L NaCl)。标准蛋白为:Ovalbumin(4.3 ×104),Conalbumin(7.5 ×104),Conalbumin(1.58 ×105),Ferritin(4.4×105),Thyroglobulin(6.69×105)。根据标准蛋白与目的蛋白的保留体积推算AspER的相对分子质量。
1.2.6 pH和温度对酶活的影响
通过测定不同pH缓冲液中AspER的酶活,确定该蛋白的最适pH。采用的缓冲液分别为柠檬酸-磷酸缓冲液(pH 2.2~8.0)和Tris-HCl缓冲液(pH 7.2~9.4)。最适反应温度则是通过测定不同温度下的酶活确定,温度范围为25~45℃,5℃为一个间隔。为了探究其温度稳定性,一定浓度的蛋白溶液在不同温度下孵育100 min,每20 min取样1次(第一次取样时孵育10 min),检测蛋白在25℃下的残余酶活力(以孵育前的酶活力为100%)。所有测定方法均按照1.2.4标准酶活测定条件测定上述不同条件的酶活,每个反应3个平行。
1.2.7 底物谱分析
按照1.2.4酶活测定方法对实验室现有的α,β-不饱和羰基等化合物进行酶活性检测以探究AspER的底物谱。考虑到底物水溶性差,所有的底物均溶解到二甲基亚砜(DMSO)体积分数1%中,其他成分溶解在pH 7.0磷酸盐缓冲液。
1.2.8 底物转化率和产物 e.e.(或 d.e.)值的测定
AspER对有活性的手性底物的立体选择性的分析采用葡萄糖脱氢酶耦联的辅酶再生体系进行。1 mL反应体系(100 mmol/L磷酸盐缓冲液,pH 7.0)中包含:葡萄糖9 mg,D-葡萄糖脱氢酶1 mg,β-NADP 1 mg,AspER 150 μg,底物 2 mg。设置的反应在37 ℃、200 r/min的条件下反应24 h后用相同体积的甲基叔丁基醚萃取,所得有机相用无水硫酸钠干燥后气相色谱仪检测底物转化率和产物 e.e.(或 d.e.)值。
2 结果和讨论
2.1 AspER的表达和纯化
考虑到蛋白可溶表达及纯化的需要,在载体构建过程中采用了pET32a(+)上的His-tag和Trx-tag。AspER,在E.coli BL21(DE3)中以可溶形式表达,经Ni-NTA纯化,蛋白纯度提高 1.9倍,回收率为60.62%;洗脱后的蛋白经肠激酶切除N端融合表达的标签后,总活力提高到53.265 U(表1),比纯化前和纯化后未切除N端标签时都高,因此认为蛋白N端融合表达的标签影响了蛋白的活性[14]。根据蛋白的氨基酸残基数预测蛋白单体大小为47.1×103,与SDS-PAGE检测结果(图1)近似。根据分子筛凝胶层析结果推算,AspER的相对分子质量为9.1×104,由此可推断该酶以二聚体形式存在。当以NADPH作为辅酶时,酶的活力约为NADH作为辅酶时酶活力的8倍,说明AspER为依赖于NADPH的氧化还原酶。
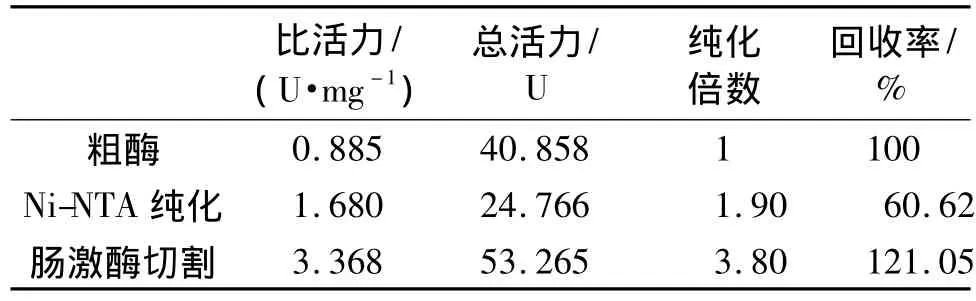
表1 AspER的纯化结果Table 1 Purification of AspER
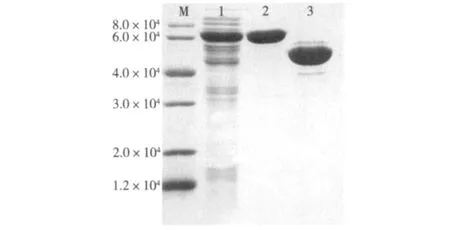
图1 AspER的SDS-PAGE检测结果Fig.1 SDS-PAGE of AspER
2.2 酶的最适反应pH、温度和温度稳定性
图2为AspER酶的最适反应pH、温度及温度稳定性结果。由图2可知:以2-环己烯酮为底物,AspER在pH 7.0~8.0时表现出了最高活性,低于pH 5.0的条件下酶完全失活(图2(a))。酶的最适反应温度及温度稳定性方面,AspER在40℃表现出了最高活性(图2(b)),在25~40℃范围孵育100 min后酶活稳定,而在45℃ 孵育10 min后酶活失去将近40%,40 min后酶基本完全失活(图2(c))。
2.3 酶促动力学参数测定
根据双倒数图(图3)计算AspER对2-环己烯酮的Km和kcat分别为(2.45±0.36)mmol/L和(4.4±0.4)×103s-1。文献报道已知烯酮/烯酯还原酶对2-环己烯酮的 Km为 0.01 ~ 5.5 mmol/L[15-19]。AspER对2-环己烯酮的Km和kcat与来源于Lactobacillis casei str.Zhang 的烯酮/烯酯还原酶(LacER)相近,LacER是NADH依赖型,而AspER更倾向于以NADPH为辅酶[22]。
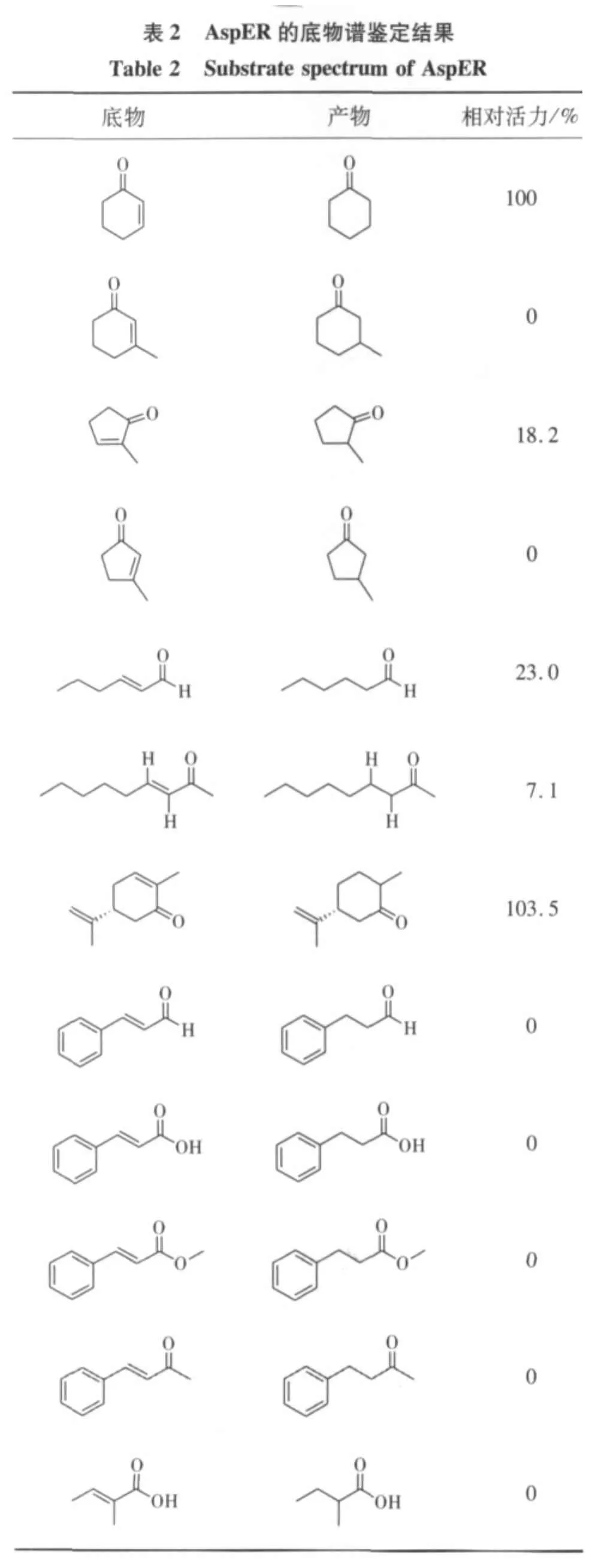
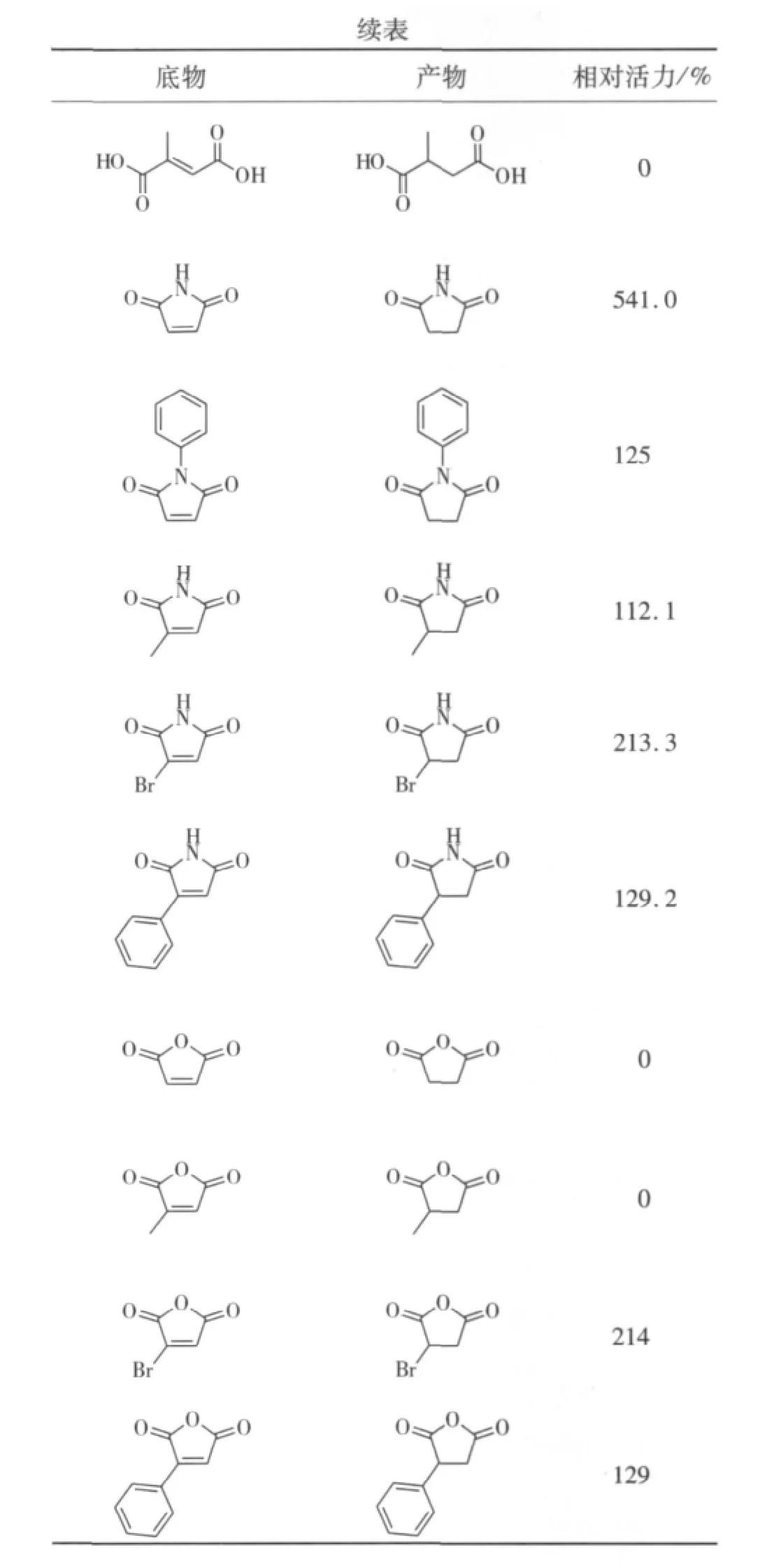
2.4 酶的底物特异性
用纯化后的AspER对不同种类的α,β-不饱和羰基化合物(醛类、环酮类、马来酰亚胺及其衍生物、羧酸和酯等)进行了底物谱筛选。以AspER对2-环己烯酮的比活定义为100,计算该酶对所有底物的相对酶活(表2)。由表2可知:AspER可以催化环状烯酮的C=C双键的还原,环上取代基的位置对催化活性有很大的影响,如2-甲基环戊烯酮可以被还原,而对3-甲基环戊烯酮则没有活性。该酶对脂肪族的醛和酮有催化活性,但不能催化苯基取代的α,β-不饱和醛、酮、羧酸及其酯的C=C双键的还原。AspER对马来酰亚胺及其衍生物有较高的活性,取代基的位置及大小对酶活有不同程度的影响。AspER对溴代马来酰亚胺和溴代马来酸酐,以及对苯基马来酰亚胺和苯基马来顺酐有相似的活性;但对马来酰亚胺和2-甲基马来酰亚胺都有较高的活性,而对马来酸酐和2-甲基马来酸酐却没有活性。至于AspER对马来酰亚胺和马来酸酐表现出如此有趣现象的原因,目前还不清楚。
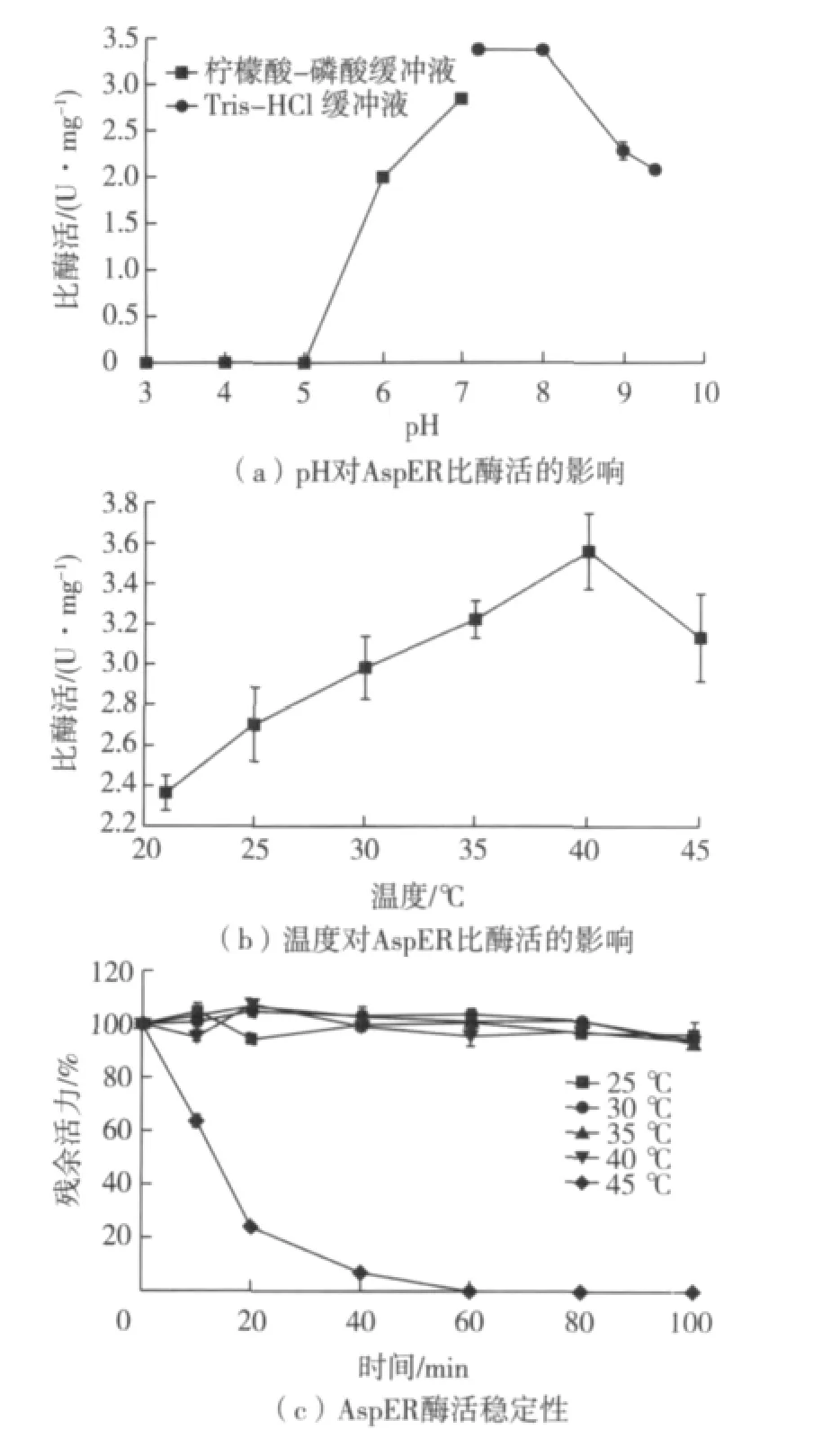
图2 pH和温度对AspER酶活的影响及酶活稳定性(以2-环己烯酮为底物)Fig.2 Effects of pH and temperature on AspER enzyme activities and its stabilities
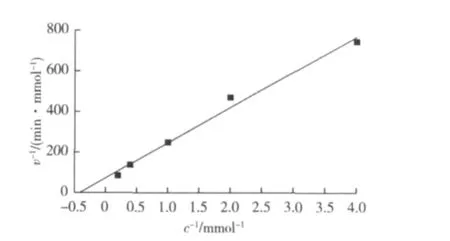
图3 双倒数作图法计算AspER酶促动力学参数Fig.3 Lineweaver-Burk plot of AspER
?
?
2.5 转化率和 e.e.(或 d.e.)值分析
用AspER对少数几个有活性的α,β-不饱和羰基化合物做了催化反应,以标准底物和消旋体产物为对照,用气相色谱仪检测了它们的转化率和产物的 e.e.(或 d.e.)值[20](表 3)。由于 2 - 甲基环戊烯酮产物的消旋体保留时间接近,不能将它们分开,所以无法确定底物转化率和产物e.e.值;由于没有溴代马来酸酐的消旋体,所以没有检测溴代马来酸酐的反应结果。由表3看出:AspER对2-甲基马来酰亚胺具有较高的转化率和立体选择性(e.e.值均高于99%);对R-香芹酮的转化率高于S-香芹酮,但具有近似的立体选择性;对2-苯基马来酰亚胺的转化率虽然较高,但立体选择性较低。
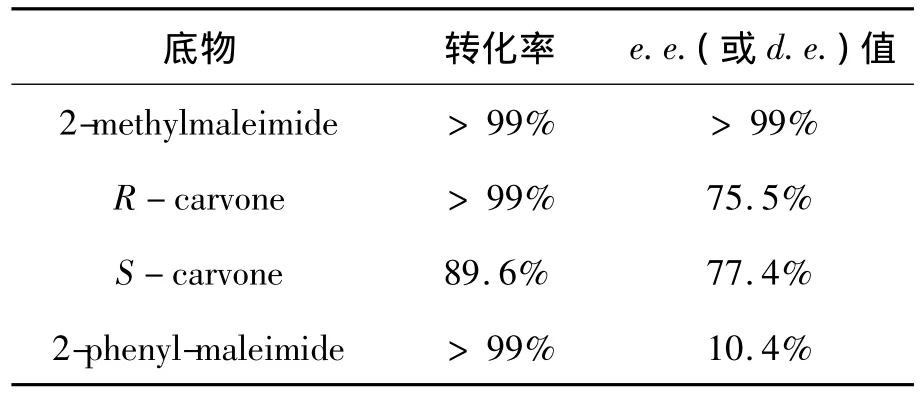
表3 AspER催化还原反应的立体选择性Table 3 Stereoselectivity of bioreduction catalyzed by AspER
3 结论
以米曲霉(Aspergillus oryzae)RIB40基因组DNA为模版,成功克隆、表达和分离纯化出一种烯酮/烯酯还原酶AspER。该酶可以利用 NADH和NADPH作为辅酶,但对NADPH有较高的偏好性。AspER对马来酰亚胺及其衍生物表现出较高的催化活性,催化2-甲基马来酰亚胺的高效立体选择性还原(转化率和e.e.值均高于99%),目前烯酮/烯酯还原酶催化马来酰亚胺及其衍生物的还原的研究还很少,AspER可望应用于该类化合物的不对称还原。
[1]Tuttle J B,Ouellet S G,David W.Organocatalytic transfer hydrogenation of cyclic enones[J].J Am Chem Soc,2006,128(39):12662-12663.
[2]Ouellet S G,Tuttle J B,David W.Enantioselective organocatalytic hydride reduction[J].J Am Chem Soc,2005,127(1):32-33.
[3]Williams R,Bruce N.New uses for an old enzyme-the old yellow enzyme family of flavoenzymes[J].Microbiology,2002,148(6):1607-1614.
[4]Swiderska M A,Stewart J D.Asymmetric bioreductions of β-nitro acrylates as a route to chiral β 2-amino acids[J].Org Lett,2006,8(26):6131-6133.
[5]Wada M,Yoshizumi A,Noda Y,et al.Production of a doubly chiral compound,(4R,6R)-4-hydroxy-2,2,6-trimethylcyclohexanone,by two-step enzymatic asymmetric reduction[J]. Appl Environ Microbiol,2003,69(2):933.
[6]Kataoka M,Kotaka A,Hasegawa A,et al.Old yellow enzyme from Candida macedoniensis catalyzes the stereospecific reduction of the C =C bond of ketoisophorone[J].Biosci Biotechnol Biochem,2002,66(12):2651-2657.
[7]Ohta H,Kobayashi N,Ozaki K.Asymmetric reduction of nitro olefins by fermenting bakers'yeast[J].J Org Chem,1989,54(8):1802-1804.
[8]Müller A,Hauer B,Rosche B.Enzymatic reduction of the α,βunsaturated carbon bond in citral[J].J Mol Catal B:Enzymatic,2006,38(3/4/5/6):126-130.
[9]Hall M,Hauer B,Stuermer R,et al.Asymmetric whole-cell bioreduction ofan [alpha],[beta]-unsaturated aldehyde(citral):co mpeting prim-alcohol dehydrogenase and C =C lyaseactivities[J].Tetrahedron:Asymmetry,2006,17(21):3058-3062.
[10]Mangan D,Miskelly I,Moody T S.A three-enzyme system involving an ene-reductase for generating valuable chiral building blocks[J].Adv Syn Catal,2012,354(11/12):2185-2190.
[11]Brenna E,Gatti F G,Manfredi A,et al.Enoate reductase mediated preparation of(S)-methyl 2-bromobutanoate,a useful key intermediate for the synthesis of chiral active pharmaceutical ingredients[J].Org Proc Res Dev,2012,16(2):262-268.
[12]Yanto Y,Winkler C K,Lohr S,et al.Asymmetric bioreduction of alkenes using ene-reductases YersER and KYE1 and effects of organic solvents[J].Org Let,2011,13(10):2540-2543.
[13]Mueller N J,Stueckler C,Hauer B,et al.The substrate spectra of pentaerythritol tetranitrate reductase,morphinone reductase,N-ethylmaleimide reductase and estrogen-binding protein in the asymmetric bioreduction of activated alkenes[J].Adv Syn Catal,2010,352(2/3):387-394.
[14]翟光磊,钟良玮.N-末端标签对人硫氧还蛋白反应特点的影响[J].生物物理学报,2009,25(增刊1):371-372.
[15]Chaparro-Riggers J,T Rogers,E Vazquez-Figueroa,etal.Comparison of three enoate reductases and their potential use for biotransformation[J].Adv Syn Catal,2007,349:1521-1531.
[16]Fitzpatric,K T,NAmrhein,Macheroux P.Characterization of YqjM,an old yellow enzyme homolog from Bacillus subtilis involved in the oxidative stress response[J].J Biol Chem,2003,278:19891-19897.
[17]French C E,Bruce N C.Purification and characterization 484 of morphinone reductase from Pseudomonas putida M10[J].Biochem J,1994,301:97-103.
[18]French C,Nicklin S,Bruce N C.Sequence and properties of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2[J].J Bacteriol,1996,178:6623-6627.
[19]Brown B J,Z Deng,P A Karplus,et al.On the active site of old yellow enzyme[J].J Biol Chem,1998,273:32753.
[20]Gao X,Ren J,Wu Q,et al.Biochemical characterization and substrate profiling of a new NADH-dependent enoate reductase from Lactobacillus casei[J].Enzyme Microb Technol,2012,51:26-34.
猜你喜欢
昆明医科大学学报(2021年5期)2021-07-22
民用飞机设计与研究(2020年1期)2020-05-21
小学生学习指导(低年级)(2017年11期)2017-10-23
中学生(2017年13期)2017-06-15
中国塑料(2017年2期)2017-05-17
中国塑料(2015年6期)2015-11-13
传奇故事(破茧成蝶)(2015年1期)2015-02-28
中国酿造(2014年9期)2014-03-11
郑州大学学报(理学版)(2014年4期)2014-03-01
无机化学学报(2014年12期)2014-02-28
