
固相萃取-超高效液相色谱-串联质谱法检测猪肉中玉米赤霉醇类的残留量
2013-10-22 11:23:06李丽萍吴国华
色谱 2013年7期
李丽萍, 范 赛, 赵 榕, 李 兵, 刘 伟, 吴国华
(北京市疾病预防控制中心营养与食品卫生所,北京 100013)
α-玉米赤霉醇(ZER)属于雷索酸内酯类(RALs)物质,同类物质还包括β-玉米赤霉醇(β-ZAL)、玉米赤霉酮(ZAN)、玉米赤霉烯酮(ZEN)、α-玉米赤霉烯醇(α-ZOL)和 β-玉米赤霉烯醇(β-ZOL),其结构相似[1-3]。RALs 物质是非甾体类雌性激素,ZER作为家畜增重的外源激素效果良好,被广泛用于动植物的促生长。20世纪70年代欧美国家就已将玉米赤霉醇作为牛羊增重剂,80年代传入我国。有数据表明雷索酸内酯类是致畸和致癌物,ZER在动物组织残留会引起人体性机能紊乱及影响第二性征的正常发育,其排出体外后还可经水和食物造成二次污染和环境污染[4-6]。ZEN具有免疫毒性、肝毒性,对肿瘤的发生也有一定影响,其广泛存在于谷物及其他农作物中,极易被用于饲料而进入畜产品体内造成残留,进而对人体健康造成威胁[7,8]。为了确保人类健康,我国、欧盟以及世界粮农组织先后明确禁止将激素类物质应用于家畜生产[8-11]。但由于玉米赤霉醇作为畜禽产品促生长剂的效果明显,部分违法者仍在畜禽养殖过程中使用玉米赤霉醇,导致玉米赤霉醇可能会残留在各种食用组织中。因此,研究和开发动物源性食品中玉米赤霉醇类药物的分析技术,对于食品安全检测具有重要意义。
目前,针对玉米赤霉醇类物质的检测方法主要有酶联免疫法(ELISA)、薄层色谱法(TLC)[12-14]、高效液相色谱法(HPLC)[15,16]、气相色谱-质谱法(GC-MS)[17,18]、液相色谱-串联质谱法 (LC-MS/MS)[19-23]等。免疫技术或酶联免疫技术虽然灵敏度高,简便快速,但假阳性率不可避免,一般用于样品筛选;气相色谱-质谱联用法通常需要复杂的衍生步骤使待测物转化为气相色谱可分析的化合物,液相色谱-质谱联用法则需要对样品进行较彻底的净化以改善待测物在质谱上的基质抑制效应。所用样品前处理技术多为液-液萃取,操作繁琐且需要使用大量的有机溶剂反复萃取。本文通过HLB小柱固相萃取进行富集净化和超高效液相色谱-串联质谱技术(UPLC-MS/MS)进行分离检测,方法简便快速、可节省大量溶剂,对环境友好,可以满足对市场上动物源性食品中痕量玉米赤霉醇类物质的监管需求。
1 实验部分
1.1 仪器与试剂
Waters ACQUITYTM超高效液相色谱仪、MICROMASSTM-Quattro premier XE质谱仪(美国Waters公司)。T25 Basic型均质器(德国 IKA公司)。Oasis HLB柱(150 mg,6 mL),ENVI-Carb GCB固相萃取小柱(500 mg,6 mL;美国Supelco公司),ENVI-Carb-NH2GCB固相萃取小柱(500 mg,6 mL;美国Supelco公司),HPLC级甲醇、乙腈(Dikma公司)。
α-玉米赤霉醇、β-玉米赤霉醇、α-玉米赤霉烯醇、β-玉米赤霉烯醇、玉米赤霉酮和玉米赤霉烯酮标准品,同位素内标α-玉米赤霉烯醇-D7、β-玉米赤霉烯醇-D7均购自加拿大TRC公司。β-葡萄糖醛酸酶/芳香基硫酸酯酶购自美国罗氏公司。
1.2 标准溶液的配制
精确称取各标准品0.0100 g,分别置于10 mL棕色容量瓶中,用甲醇溶解并定容至10 mL,配成质量浓度为1000 mg/L的标准储备液,于-20℃冰箱保存。需要时各取储备液0.1 mL置于10 mL棕色容量瓶中,并用甲醇稀释成10 mg/L的混合中间液,使用前将中间液以甲醇稀释配制成1.0 mg/L的混合标准液。同位素内标α-玉米赤霉烯醇-D7和β-玉米赤霉烯醇-D7单独配制成10.0 mg/L的标准中间溶液,使用前将中间液以甲醇稀释配制成1.0 mg/L的混合标准液作为标准使用液。两种溶液均于避光、-20℃下保存。
1.3 样品前处理
准确称取5.0 g均质猪肉样品,加入20 μL的1.0 mg/L内标溶液和10 mL pH 5.2的乙酸-乙酸钠缓冲溶液,涡旋混匀、超声30 min,再加入β-葡萄糖醛酸酶/芳香基硫酸酯酶溶液100 μL,于(37±1)℃振荡酶解12 h,取出后冷却至室温,加入25 mL甲醇超声提取30 min,0~4℃下以10000 r/min离心10 min,转移上清液至另一洁净试管中,待净化。Oasis HLB柱(6 mL,150 mg)依次用6 mL甲醇、6 mL水活化,提取液上柱后,用3 mL超纯水淋洗小柱,减压抽干。以6 mL甲醇洗脱,收集洗脱液,在40℃下氮气浓缩至干,残留物用初始流动相复溶后过0.22 μm的有机滤膜供UPLC-MS/MS测定。
1.4 色谱-质谱条件
1.4.1 液相色谱条件
色谱柱:Waters ACQUITY UPLCTMBEH C18柱(100 mm ×2.1 mm,1.7 μm);柱温:40 ℃;样品室温度:4℃;进样体积:5 μL。流动相A为水,流动相B为乙腈;梯度洗脱程序:0~5 min,65%A线性降到40%A;5~5.10 min,40%A线性降到5%A,并保持到7.50 min;7.50~8.00 min,5%A降到0%A,并保持到8.50 min;8.50~9.00 min,0%A线性升到 65%A,并保持到 10.0 min。流速为 0.3 mL/min。
1.4.2 质谱条件
电离方式:电喷雾负离子模式(ESI-);多反应监测模式(MRM)采集,驻留时间0.10 s;毛细管电压:2.70 kV;锥孔电压:45 V;离子源温度:100℃;脱溶剂温度:400℃;脱溶剂气流量:600 L/h;碰撞室压力:0.33 Pa。其他质谱条件见表1。
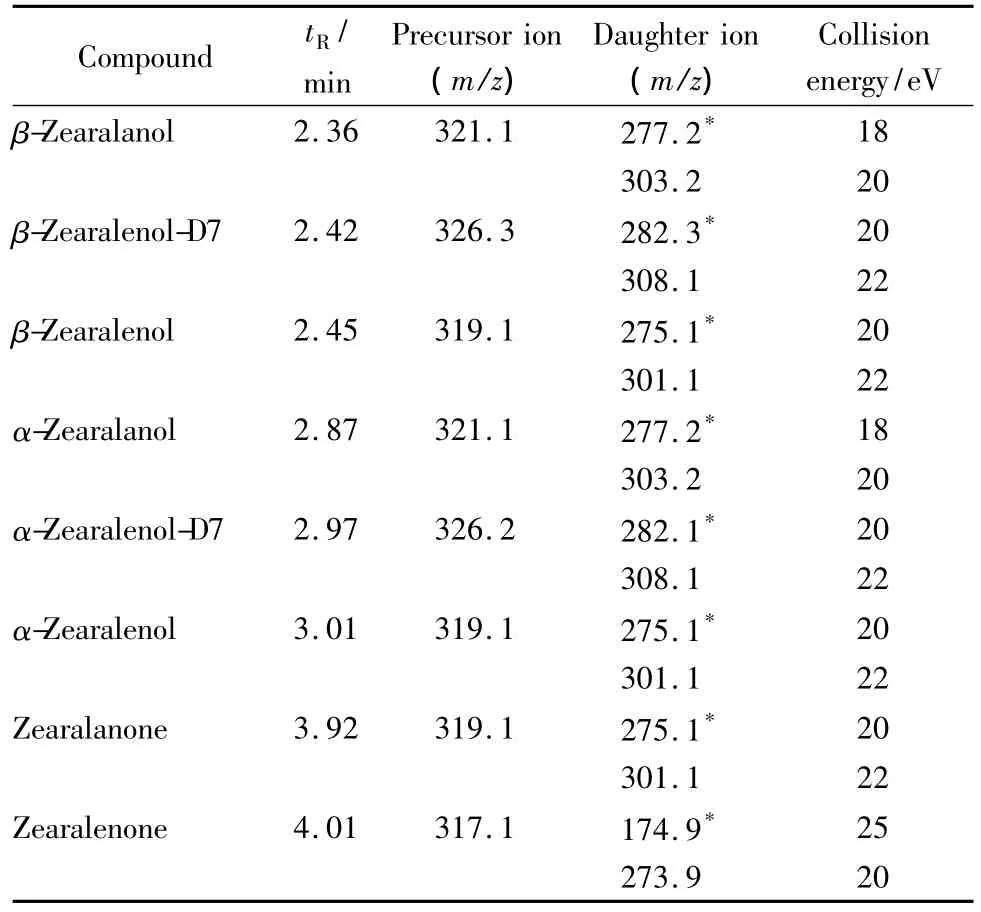
表1 多反应监测模式下6种化合物的检测参数Table 1 MRM parameters of the six compounds
2 结果与讨论
2.1 仪器条件的优化
通过流动注射泵连续进样200 μg/L以甲醇-水溶液(50∶50,v/v)为溶剂的单标准溶液,并同时开启液相色谱,设定其流动相为甲醇-水溶液(50∶50,v/v),流速为50 μL/min。在此条件下对质谱参数如毛细管电压、锥孔电压、脱溶剂气流速等参数进行了正、负离子优化,优化结果见1.4.2节。玉米赤霉醇及其类似物均属于二羟基苯甲酸内酯类化合物,其苯环上都有2个羟基,呈弱酸性。在液相色谱-电喷雾质谱检测时易失去质子而产生[M-H]-准分子离子。经过优化后,结果表明6种玉米赤霉醇类物质均在负离子模式下产生[M-H]-的最强准分子离子峰。从理论上推测,偏碱性的流动相更易提高ESI-模式下目标化合物的响应。因此,本研究考察了分别以0.1%氨水溶液-甲醇、10 mmol/L乙酸铵水溶液-甲醇、纯水-甲醇、0.1%甲酸水溶液-甲醇、0.1%氨水溶液-乙腈、10 mmol/L乙酸铵水溶液-乙腈、纯水-乙腈、0.1%甲酸水溶液-乙腈作为流动相时对目标化合物色谱行为与质谱响应的影响。但结果显示,以纯水和乙腈为流动相时,6种目标物质分离完全,灵敏度最高。因此,本研究最终选择水和乙腈为流动相。
2.2 样品前处理条件的选择
玉米赤霉醇类化合物是一种类雌激素物质,在动物组织中会以硫酸轭合物或葡萄糖醛酸轭合物等形式存在,因此样品要先进行水解,使待测物游离或从组织中释放以进行后续的提取工作。目前,对于动物源性食品中玉米赤霉醇类物质的提取方式国内外均有报道,但多集中于使用乙醚、丙酮、正己烷和乙酸乙酯等极性相对较弱的提取液进行提取,经NaOH溶液反萃后,再用反相固相萃取柱净化[24]。方法步骤繁琐,且耗费大量的溶剂。如农业部1025号公告-19-2008和GB/T 21982-2008,二者在检测手段上与本文相差不大,但前处理过程则较为复杂[23,24]。农业部 1025 号公告-19-2008 中酶解后首先采用正己烷反复萃取,旋蒸至近干后再用乙酸乙酯复溶才能过SPE小柱净化;而GB/T 21982-2008中酶解后首先采用乙醚反复萃取,旋蒸至近干后再用二氯甲烷复溶,还要经NaOH溶液反萃才能过SPE小柱净化。而对植物性食品中玉米赤霉醇类物质的报道则多集中于使用84%乙腈水溶液进行提取[19]。考虑到玉米赤霉醇类物质的物理化学性质类似,因此本实验尝试采用了甲醇水溶液作为提取液进行提取,避免了采用正己烷或乙醚提取而产生的脂类杂质的干扰,省去了采用NaOH溶液反萃的去脂过程,从而减少了提取步骤,并优化了提取效率,避免了反复转移所带来的目标物损失,净化效果对比如图1所示,低水平加标的离子流图如图2所示。并且由于精简了提取过程,也减少了大量有机溶剂的使用和工作时间,对环境友好,更适用于大量样品的监测工作。通过对酶解后的溶液分别加入10、15、20、25、30 mL 的甲醇进行提取,并搭配 ENVICarb固相萃取柱、NH2固相萃取柱、ENVI-Carb固相萃取柱串接NH2固相萃取柱、HLB固相萃取柱进行对比试验。结果显示,HLB固相萃取柱能同时满足6种玉米赤霉醇类化合物的净化要求,效果最佳。HLB柱在20 μg/kg加标水平下,不采用内标法计算的绝对回收率均可以达到80%以上,其他两种小柱或两种小柱的组合其绝对回收率只有30%~50%,且部分化合物没有检出。因此本研究最终选定HLB柱作为净化手段。
2.3 方法的线性关系、检出限与定量限
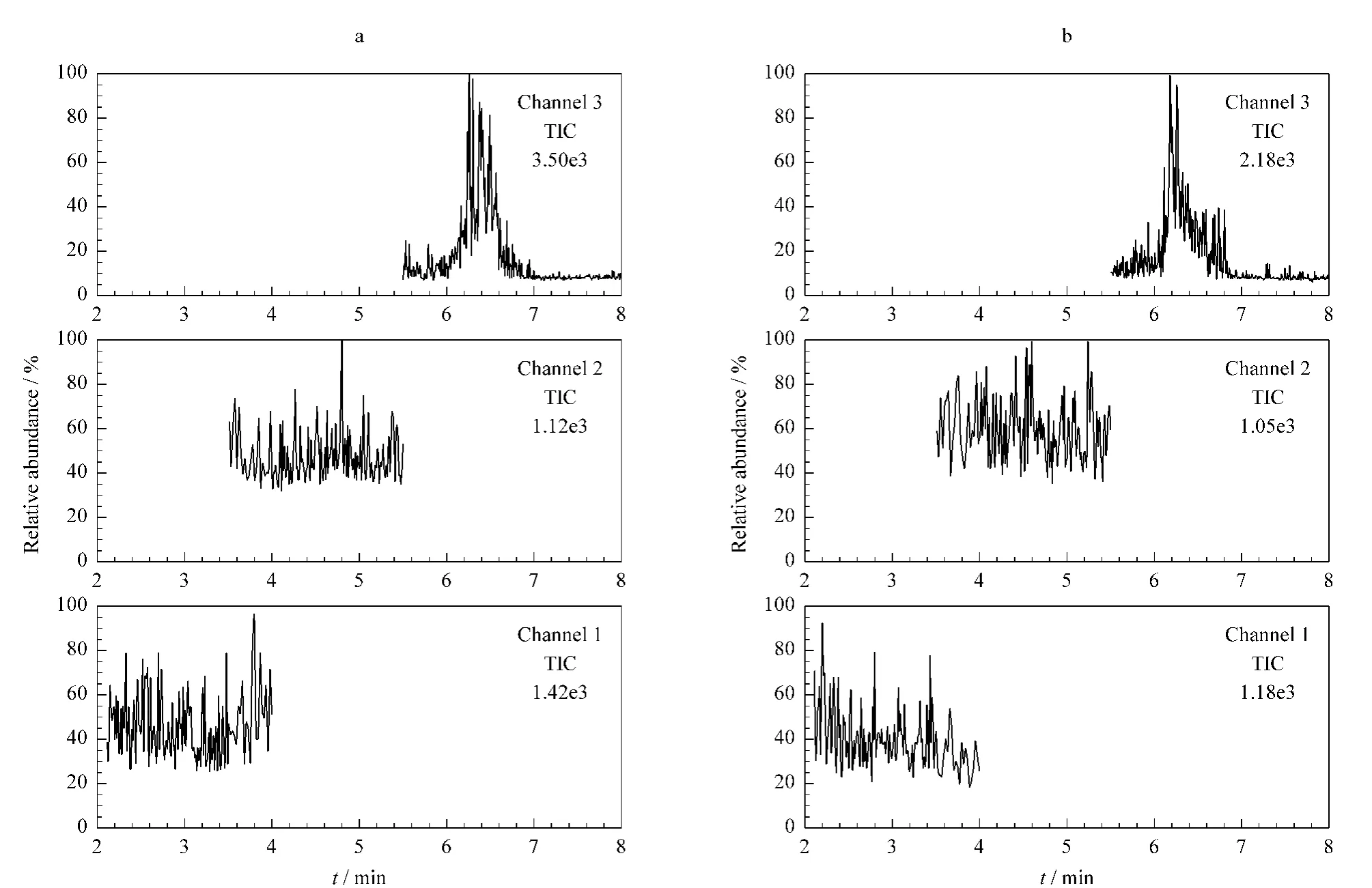
图1 采用(a)国标方法与(b)本方法对空白样品的净化效果对比Fig.1 Comparison of purification effects of the blank sample by(a)the GB method with(b)this method
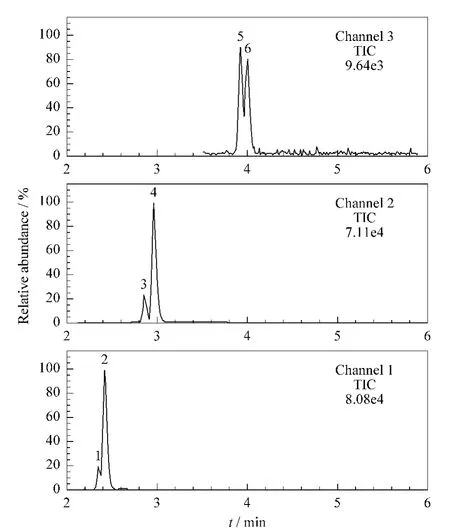
图2 6种化合物的加标样品的总离子流图(0.1 μg/kg)Fig.2 Total ion chromatograms of the six standards fortified in a blank sample at the level of 0.1 μg/kg
将标准使用液用甲醇-水(50∶50,v/v)溶液作溶剂逐级稀释,配制成质量浓度分别为1.0、5.0、10.0、25.0、50.0、100.0 μg/L 的系列混合标准溶液,其中均含有5 μg/L的内标。并采用液相色谱-质谱联用仪进行分析,记录各化合物的色谱峰面积(A)和内标的色谱峰面积(Ai),以A和Ai的比值y(氘代同位素内标除对应各自的化合物外,其他化合物按色谱保留时间顺序以相近的氘代同位素化合物为内标)对相应的标准溶液中化合物的质量浓度x进行线性回归计算,得出线性方程、线性范围和相关系数(r),结果见表2。由结果可见,在1.0~100.0 μg/L范围内,各物质均取得良好线性相关,r大于0.999。质量浓度为10.0 μg/L的标准溶液的MRM的提取离子色谱图见图3。样品中目标化合物的色谱峰与相应标准品的色谱峰的保留时间必须一致,每种化合物的两对离子必须同时出现,且丰度比符合欧盟关于食品中污染物的确认依据2002/657/EC[25]。待测化合物的定性离子色谱峰的信噪比(S/N)应不小于3,定量离子色谱峰的S/N应不小于10。
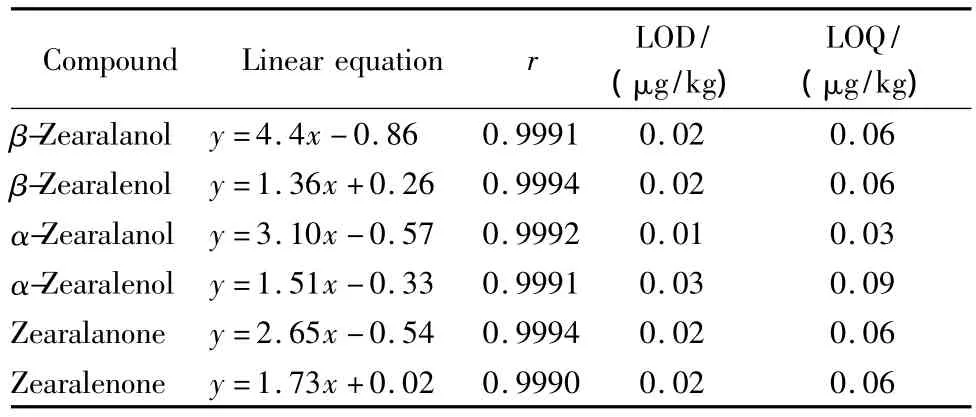
表2 各化合物的线性方程、相关系数、检出限和定量限Table 2 Linear equations,correlation coefficients(r),LODs and LOQs of the six compounds

图3 6种目标化合物标准溶液(含2种内标)的MRM色谱图Fig.3 MRM chromatograms of a standard mixture of the six compounds and two internal standards
2.4 方法的回收率和精密度
通过向阴性猪肉样品基质中添加分析物进行测定,以确定方法的准确度和精密度。首先精确称取5.0 g猪肉样品,加入适量的混合标准溶液,使得玉米赤霉醇类物质的含量分别达到1.0、5.0、10.0 μg/kg,按优化的条件进行处理和测定,采用内标法进行计算,结果见表3。本方法灵敏度高,定性准确,可以用于猪肉中玉米赤霉醇类药物残留的确证检测。
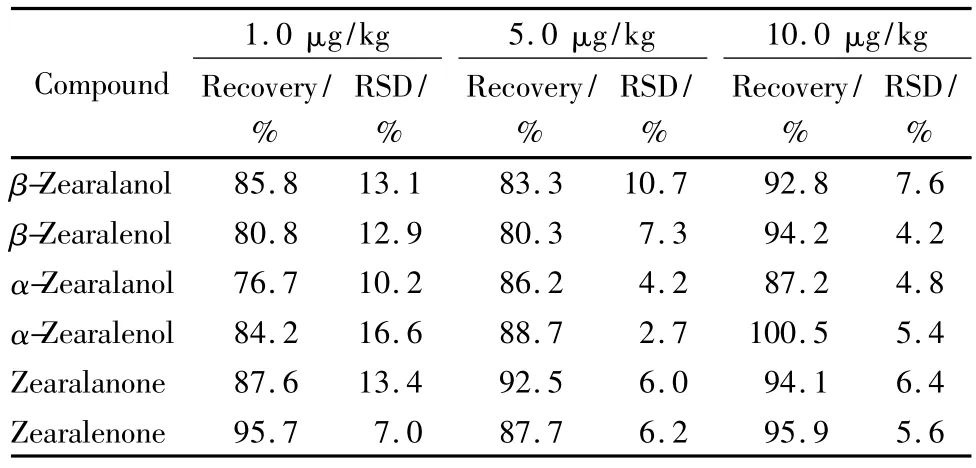
表3 3个加标水平下的回收率试验结果(n=6)Table 3 Results of the recovery test at three spiked levels(n=6)
2.5 样品测定
通过采用所建立的方法,对所采集的30份猪肉样品进行了检测,但在采集的样品中均未检出上述6种玉米赤霉醇类物质。同时进行的作为质量控制数据的加标回收试验结果也满足测定结果的要求,回收率在70%以上。计划再次采集大量样品进行检测以确证方法的适用性。
3 结论
本文采用酶解方式对猪肉样品进行彻底水解,采用HLB固相萃取柱净化,以超高效液相色谱-串联质谱对6种玉米赤霉醇类物质进行检测。本方法准确可靠、灵敏度度高,适用于动物源性食品中玉米赤霉醇类物质的多残留确证和定量检测。
[1]Kennedy D G,Hewitt S A,McEvoy J D G,et al.Food Addit Contam,1998,15(4):393
[2]Wang S,Wang X H.Food Addit Contam,2007,24(6):573
[3]Kleinova M,Zollner P,Kahlbacher H,et al.J Agric Food Chem,2002,50(17):4769
[4]Richardson K E,Hagler W M J,Mirocha C J.J Agric Food Chem,1985,33(5):862
[5]Dong M,He X J,Tulayakul P,et al.Toxicon,2010,55(2/3):523
[6]Tsakmakidis I A,Lymberopoulos A G,Khalifa T A A,et al.J Appl Toxicol,2008,28(5):681
[7]Draughon F A,Churchville D C.Phytopathology,1985,75(5):553
[8]Joint FAO/WHO Expert Committee on Food Additives(JECFA).WHO Food Additives Series.Geneva:World Health Organization,2000:44
[9]Bulletin No.235,Ministry of Agriculture,PR China.Maximum Residue Limits of Veterinary Drugs in Food of Animal Origin(中华人民共和国农业部第235号公告:动物源性食品中兽药最高残留限量).2002:Appendix 4
[10]The Council of the European Union.Council Directive 96/22/EC of 29 April 1996 Concerning the Prohibition on the Use in Stockfarming of Certain Substances Having Hormonal or Thyrostaic Action and of Beta-agonists and Repealing Directives 81/602/EEC,88/146/EEC and 88/299/EC.Off J Eur Comm,L125:3
[11]Food and Agriculture Organization of the United Nations(FAO).Worldwide Regulations for Mycotoxins in Food and Feed in 2003.Rome,2004.[2012-12-18].http://www.fao.org/docrep/007/y5499e/y5499e00.htm
[12]Sydenham E W,Gelderblom W C A,Thiel P G,et al.J Agric Food Chem,1990,38(1):285
[13]Burmistrova N A,Goryacheva I Y,Basova E Y,et al.Anal Bioanal Chem,2009,395(5):1301
[14]Medina M B,Nagdy N.J Chromatogr B,1993,614(2):315
[15]Zeng H Y,Li Y Q,Jing H Q.Chinese Journal of Analytical Chemistry(曾红燕,黎源倩,敬海泉.分析化学),2006,34(3):351
[16]Cavaliere C,D'Ascenzo G,Foglia P,et al.Food Chem,2005,92(3):559
[17]You L N,Li X L,Xi C X,et al.Chinese Journal of Chromatography(游丽娜,李贤良,郗存显,等.色谱),2012,30(10):1021
[18]Zhang W,Wang J P,Shen J Z,et al.Acta Veterinaria etZootechnica Sinica(张伟,王建平,沈建忠,等.畜牧兽医学报),2007,38(5):513
[19]Meng J,Zhang J,Zhang N,et al.Chinese Journal of Chromatography(孟娟,张晶,张楠,等.色谱),2010,28(6):601
[20]Hartmann N,Erbs M,Wettstein F E,et al.J Chromatogr A,2007,1138(1/2):132
[21]Xia X,Li X W,Ding S Y,et al.J Chromatogr A,2009,1216(12):2587
[22]Ren Y,Zhang Y,Shao S,et al.J Chromatogr A,2007,1143(1/2):48
[23]Bulletin No.1025,Ministry of Agriculture,PR China(中华人民共和国农业部第1025号公告).2008
[24]GB/T 21982-2008
[25]Commission Decision 2002/657/EC of12 August2002.Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results(2002).Off J Eur Comm,L221:8
猜你喜欢
能源化工(2022年3期)2023-01-15 02:26:43
煤化工(2022年3期)2022-07-08 07:24:42
中国饲料(2022年5期)2022-04-26 13:42:46
食品安全导刊·中旬刊(2022年3期)2022-04-15 22:07:47
河北果树(2022年1期)2022-02-16 00:41:06
环境与发展(2019年10期)2019-11-28 14:25:24
课程教育研究·学法教法研究(2019年10期)2019-05-14 11:02:18
中国粮油学报(2016年1期)2016-02-06 02:17:05
中国资源综合利用(2016年10期)2016-01-22 08:36:09
汽车文摘(2014年8期)2014-12-16 01:54:28
