
气相催化脱氟化氢制备含氟烯烃催化剂的研究进展
2013-10-11 02:51白彦波马洋博
化工进展 2013年10期
白彦波,马洋博,毛 伟,秦 越,吕 剑
(西安近代化学研究所,陕西 西安 710065)
含氟烯烃的制备具有重要的经济和环境价值。含氟烯烃可用作许多含氟聚合物的单体和含氟精细化学品的中间体,如合成聚氟乙烯的氟乙烯[1]、合成三氟溴乙烯的三氟乙烯[2]等。同时,某些含氟烯烃的全球变暖潜值极低,对全球温室效应影响小,被认为是新一代环保型制冷剂,如1,3,3,3-四氟丙烯(HFO-1234ze)、2,3,3,3-四氟丙烯(HFO-1234yf)[3-8]。含氟烯烃的制备路线较多,其中含氟烷烃脱氟化氢合成含氟烯烃是一简便、高效的方法,是氟化工界研究的热点之一。
含氟烷烃脱氟化氢制备含氟烯烃的方法有三种:高温裂解法[9-11]、液碱法[12-16]和气相催化法[17-18]。高温裂解法能耗高,反应设备要求苛刻,副产物多;液碱法需要使用强碱,副产大量盐,大规模使用会带来环境问题;气相催化法反应温度较低,目标产物选择性高,成为目前含氟烷烃脱氟化氢制备含氟烯烃的研究重点。脱氟化氢催化剂则在气相催化法的开发中居于核心地位。本文介绍了气相脱氟化氢反应及所用催化剂的研究进展,比较了各类催化剂的使用性能,以期为该领域的发展有所借鉴。
1 气相脱氟化氢反应
脱氟化氢反应是典型的卤代烃消去反应。研究表明卤代烃消去有E1、E2和E1cB三种机理[19-21]。E1机理首先是碳卤键的异裂,生成碳正离子,碳正离子再脱去质子完成反应;部分碳正离子会发生重排,生成同分异构体。E2机理是离去的卤原子和离去的氢原子相互作用,通过协同过程一步完成反应。E1cB机理首先是碳氢键的异裂,生成碳负离子,最后碳负离子脱去卤负离子完成反应。
Kamiguchi等[22]给出了典型卤代烃脱卤化氢发生消去反应的机理示意图(图1)。一般认为气相催化脱氟化氢反应是按 E1机理进行,反应的初始步骤是C—F键的活化异裂。通常氟的最强电负性使含氟烷烃中的氟原子带有部分负电荷,可以通过和催化剂的酸性位作用加速 C—F键的异裂。Teinz等[23]认为,具有较强酸性的氟化铝是有效的脱氟化氢催化剂,氟化铝的强酸中心和氟原子相互作用引发反应。Li等[24]提出1,1,1-三氟乙烷(HFC-143a)脱氟化氢生成 1,1-二氟乙烯的活性位是磷酸盐类催化剂的弱酸位,结合产物分析得出反应是通过碳正离子中间体进行的。
2 气相催化脱氟化氢催化剂
气相催化脱氟化氢催化剂可分为活性炭基催化剂、铬基催化剂、铝基催化剂及其它催化剂。
2.1 活性炭基催化剂
活性炭(AC)由于具有丰富的孔结构,较大的比表面积,可单独作为主体催化剂使用,也可用作负载型催化剂的载体。
Mukhopadhyay等[25]将 3种不同活性炭和两种活性炭负载的贵金属催化剂用于1,1,1,2,2-五氟丙烷(HFC-245cb)气相脱HF制备HFO-1234yf。负载贵金属后,反应活性有所提高,但 HFO-1234yf选择性有所下降, 0.5%Pd/AC显示出较好的性能,结果见表1。Miller等[26]也报道了相似的反应结果,以 AC为催化剂,在反应温度 400℃,N2/HFC-245cb=1.5,接触时间120s时,HFC-245cb的转化率虽然只有23.6%,但HFO-1234yf的选择性高达97%。Miller等[27]也研究了AC催化1,1,1,3,3-五氟丙烷(HFC-245fa)气相脱 HF制备 1,1,1,3-四氟丙烯(HFO-1234ze)的反应性能,在反应温度350 ℃,N2/HFC-245fa=1,接触时间75 s时,HFC-245fa的转化率为38%,HFO-1234ze的选择性达96%。
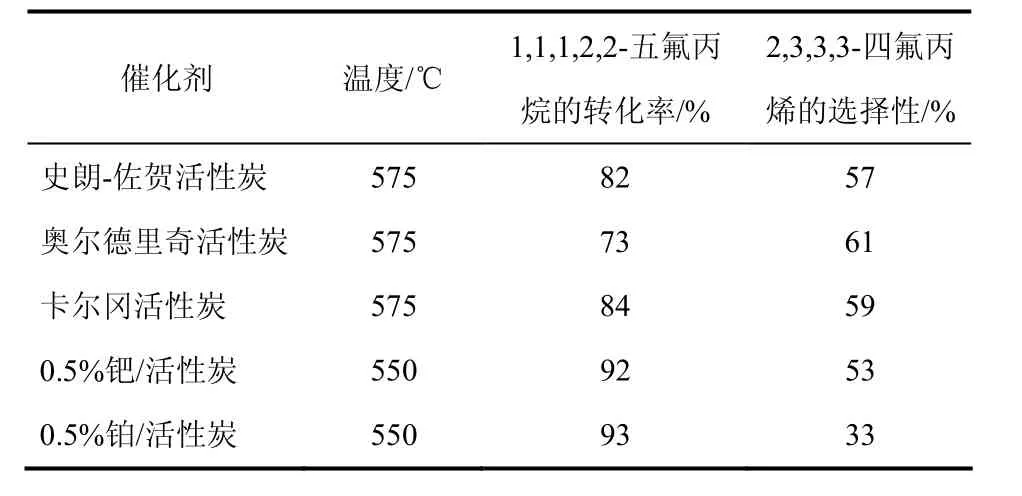
表1 用于1,1,1,2,2-五氟丙烷制备2,3,3,3-四氟丙烯的催化剂性能比较
对AC进行酸洗处理可以降低表面灰分,提高反应活性。Mukhopadhyay等[28]比较了未处理 AC和酸洗后 AC催化 HFC-245fa脱 HF制备HFO-1234ze的反应性能。在反应温度515℃时,酸洗处理可使AC的活性由88%提高到100%。Sakyu等[29]用浸渍法分别制备了 5%MoCl5/AC、5%TiCl4/AC、5%IrCl4/AC和5%SnCl4/AC四种催化剂,经氟化氢气体处理后,用于HFC-245fa脱HF制备HFO-1234ze。在反应温度350℃时,虽然各催化剂的 HFO-1234ze选择性均在 96%以上,但5%MoCl5/AC的活性最高,转化率可达58%。

Puy等[30]采用AC和0.5%Pd/AC催化1,1,1,2,3-五氟丙烷(HFC-245eb)脱HF制备HFO-1234yf。0.5%Pd/AC的性能明显优于未负载AC,结果见表2。Mukhopadhyay 等[31]将 4%~6%FeCl3/AC 用于HFC-245eb制备HFO-1234yf的反应。在反应温度300℃时,HFC-245eb的转化率可达 59%,但HFO-1234yf选择性只有37%。

表2 用于1,1,1,2,3-五氟丙烷制备2,3,3,3-四氟丙烯的催化剂性能比较
Puy等[30]采用 0.5%Fe/AC、5.0%Co/AC 和0.5%Ni/AC催化1,1,1,2,3,3-六氟丙烷(HFC-236ea)气相脱 HF制备 Z-1,1,1,2,3-五氟丙烯(Z-HFO-1225ye)。在反应温度350℃时,催化性能如 下 : 0.5%Fe/AC>0.5%Ni/AC>5.0%Co/AC 。0.5%Fe/AC用作催化剂时,HFC-236ea的转化率可达80.0%,Z-HFO-1225ye选择性为88.5%。
虽然活性炭基催化剂制备简单,所用活性炭来源广泛,成本低廉,但此类反应容易发生催化剂结焦失活,而活性炭基催化剂失活后难以再生[32]。
2.2 铬基催化剂
铬基催化剂主要组成是氧化铬、氟化铬或氟化的氧化铬,它们既可制成主体催化剂使用,又可当作负载型催化剂的载体使用。
Hirokazu等[33]分别以氟化的氧化铬和氧化铬为催化剂,考察两者催化HFC-236ea制备HFO-1225ye的反应性能,结果表明(表3),氟化后的氧化铬性能优于未经氟化的氧化铬,氟化处理可提高催化剂的反应性能。
Sakyu等[29]使用氟化的氧化铬为催化剂,考察其催化HFC-236ea制备Z-HFO-1225ye的性能。在反应温度 350℃时,HFC-236ea转化率和Z-HFO-1225ye选择性均在 94%以上,显示出氟化的氧化铬的优异脱HF性能。Nappa等[34]分别使用氟化的 α-Cr2O3和氟化的氧化铬凝胶为催化剂,用于HFC-245eb制备HFO-1234yf反应。二者均显示出较好的催化性能,HFC-245eb的转化率分别为96%和 98%,HFO-1234yf的选择性分别为 69%和71%,但氟化的氧化铬凝胶催化性能略优。Christoph等[35]以氟化的 α-Cr2O3为催化剂,用于 1,1-二氟乙烷(HFC-152a)气相脱HF制备氟乙烯反应。在反应温度350 ℃时,HFC-152a的转化率为79%,氟乙烯的选择性高达97%。Sievert[36]以氟化的氧化铬为催化剂,催化1,1,1,2,2,3-六氟丙烷(HFC-236cb)制备 HFO-1225ye。在反应温度 350 ℃时,HFC-236cb的转化率只有40%,但HFO-1225ye选择性高达90%。Nappa等[37]也报道了相似的研究成果,以氟化的氧化铬为催化剂,在反应温度348 ℃时,HFC-236cb的转化率为40.5%,HFO-1225ye的选择性达91.1%。Rao等[38]分别以立方氟化铬和斜方六面体氟化铬为催化剂催化HFC-152a制备氟乙烯,发现立方氟化铬的性能优于斜方六面体氟化铬。

表3 用于1,1,1,2,3,3-六氟丙烷制备1,1,1,2,3-五氟丙烯的催化剂性能比较
Rao等[39]用氟化的 5%Co/Cr2O3为催化剂,催化HFC-245eb制备HFO-1234yf。在反应温度250 ℃时,HFC-245eb的转化率为55%,HFO-1234yf的选择性达98%。Powell 等[40]使用3%Zn/Cr2O3为催化剂,催化1,1,1,2-四氟乙烷(HFC-134a)气相脱HF制备三氟乙烯。在反应温度500℃时,HFC-134a的转化率只有13%,但三氟乙烯的选择性高达99%。Smith等[41]使用 6%Zn/Cr2O3为催化剂,催化HFC-236ea制备HFO-1225ye。在反应温度320 ℃,接触时间5 s时,HFC-236ea的转化率为94.7%,HFO-1225ye的选择性可达97.5%。Amos等[42]使用氟化的1.9%Zn/Cr2O3为催化剂,催化1,1,1,3,3,3-六氟丙烷(HFC-236fa)制备 1,1,3,3,3-五氟丙烯(HFO-1225zc)。在反应温度 400 ℃,N2/HFC-236fa=4,接触时间15s时,HFC-236fa的转化率只有40%,但HFO-1225zc的选择性可达90%。
铬基催化剂在使用中存在失活的问题。Hirokazu等[33]发现氟化的氧化铬用于HFC-236ea制备 HFO-1225ye反应,运转 100 h后,HFC-236ea的转化率由85.1%下降到45.2%,催化剂失活严重。Hirokazu等[33]同时发现,在反应过程中通入氧气或空气等气体可延缓失活,反应运转100 h后,催化剂的性能几乎没有下降。失活的铬基催化剂可通过烧炭进行再生。Nappa等[37]在425 ℃,通入空气对失活的氟化的氧化铬催化剂进行再生,HFC-236cb的转化率可由23.8%恢复到初始的40.1%。
2.3 铝基催化剂
铝基催化剂主要组成是氟化铝或氟化的氧化铝,它们既可以制成主体催化剂使用,也可以当作负载型催化剂的载体使用。
Miller等[27]将氟化的氧化铝用于HFC-245cb制备HFO-1234yf的反应。在反应温度400 ℃,接触时间 60 s时,HFC-245cb的转化率为 98%,HFO-1234yf的选择性可达93%。Miller等[43]将氟化的氧化铝用于HFC-245fa制备HFO-1234ze的反应。在反应温度375 ℃,接触时间30 s时,HFC-245fa的转化率为 84.8%,HFO-1234ze的选择性可达98.5%。Mahler等[44]将氟化的氧化铝用于催化HFC-245eb制备HFO-1234yf。在反应温度400 ℃时,转化率和选择性均不到 70%。Uenveren等[45]分别将AlF3和 AlF3/γ-Al2O3用于催化 HFC-134a制备三氟乙烯反应。在反应温度300 ℃时,HFC-134a的转化率分别为 10.8%和 12.2%,三氟乙烯的选择性分别为89.0%和83.7%。载体γ-Al2O3具有强的酸性,使用可增强催化剂的酸性,提高反应活性,但过强的酸性会导致副产物增多,降低目标产物选择性。
MgF2的引入可改善铝基催化剂的性能。Nappa等[46]分别使用 α-AlF3、β-AlF3和 10%MgF2/AlF3为催化剂,催化HFC-152a制备氟乙烯。催化活性如下:10%MgF2/AlF3>α-AlF3>β-AlF3。Rao 等[1]分别使用 ZnF2/β-AlF3和 MgF2/β-AlF3为催化剂,催化HFC-152a制备氟乙烯。二者均显示出较好的性能,氟乙烯的选择性均为100%,运行48 h催化剂都无失活。Sakyu等[29]采用 AlF3和 10%MgF2-90%AlF3为催化剂,催化HFC-236ea制备HFO-1225ye,发现10%MgF2-90%AlF3的催化性能优于AlF3。Merkel等[47]也报道了相似的结果,在催化HFC-245fa制备HFO-1234ze的反应中,10%MgF2-90%AlF3活性更优。蔚辰刚等[2]比较了 AlF3和 MgF2/AlF3催化HFC-134a制备三氟乙烯的性能,发现 MgF2/AlF3的催化活性比 AlF3低,但选择性比 AlF3高,说明MgF2的引入可抑制催化剂的活性,提高选择性,同时改善催化剂的稳定性。
Elsheikh等[48]将 6%Ni-6%Cr/AlF3分别用于HFC-245cb和HFC-245eb脱HF制备HFO-1234yf反应,原料转化率和目标产物选择性均在70%以上。Elsheikh等[32]将 AlF3和 6%Ni/AlF3用于催化HFC-245eb制备 HFO-1234yf。结果表明,Ni的引入可提高催化剂活性,HFC-245eb的转化率由34%提高到 97%。Puy等[30]将氟化的 Zr/Al2O3用于HFC-245fa制备HFO-1234ze反应。在反应温度300℃时,HFC-245fa转化率和 HFO-1234ze选择性均在94%以上。
铝基催化剂在使用中也存在失活问题。Powell等[40]使用氟化铝为催化剂,催化HFC-134a制备三氟乙烯,发现反应进行190 min后,HFC-134a的转化率即由最初的 38.4%下降到 5.8%。Powell等[40]同时发现,氟化铝掺杂铁后,初始转化率虽有所下降,但在反应进行259 min后,转化率几乎没有下降,表明铁的加入可延缓催化剂失活。Sheppard等[49]发现,在反应中引入适量的氧气可使8.5%CrF3/AlF3的寿命由20 h增加到104 h。失活的铝基催化剂可通过烧炭进行再生。Powell等[40]在600 ℃通入空气对失活的氟化铝进行再生,使HFC-134a的转化率由失活时的5.8%恢复到22.4%。
2.4 其它催化剂
Mukhopadhyay等[25]使用Ni网为催化剂,催化HFC-245cb制备HFO-1234yf。在反应温度为550 ℃时,HFC-245cb的转化率为73%,HFO-1234yf的选择性达46%。Nappa等[46]分别将氟化锌、氟化的氧化镁、氟化钙用作催化剂,催化HFC-152a制备氟乙烯。催化活性有如下顺序:氟化的氧化镁>氟化锌>氟化钙。Sievert等[50]分别将 MgF2、MgF2/AlF3(43∶1)和 MgF2/AlF3(9∶1)用于催化 HFC-236fa制备 HFO-1225zc反应。MgF2/AlF3(43∶1)和MgF2/AlF3(9∶1)的催化性能优于MgF2,但两者的性能相近。HFO-1225zc的选择性均高达99.6%,但 MgF2/AlF3(43∶1)活性略高于 MgF2/AlF3(9∶1)。Nappa等[51]将氟化的氧化镧用于催化HFC-236ea制备HFO-1225ye。在反应温度为500 ℃时,HFC-236ea的转化率只有16%,但HFO-1225ye的选择性可达99%。
3 结 语
长期以来,催化剂积炭失活一直是制约气相催化脱氟化氢制备含氟烯烃反应发展的核心问题。活性炭基催化剂难以实现反应过程中通空气或氧气来延缓失活,同时无法通过烧炭再生,使用受到限制。铬基催化剂和铝基催化剂均可通过在反应过程中通入空气或氧气延缓失活,也可通过烧炭再生。由于铬存在众所周知的环境污染问题,导致铬基催化剂的使用不符合绿色化工的要求,减少其使用甚至完全淘汰将是今后发展的趋势。因此,铝基催化剂以性能优异、绿色环保,成为气相脱氟化氢催化剂开发的焦点之一。添加合适的助剂对铝基催化剂进行改性,进一步提高其性能是研究的一个重要方向。
[1]Rao V N,Subramanian M A.Catalytic manufacture of vinyl fluoride:US,5880315[P].1999-03-09.
[2]蔚辰刚,谢冠群,周强,等.MgF2-AlF3催化剂用于四氟乙烷裂解制备三氟乙烯[J].工业催化,2012,20(4):56-59.
[3]王博,张伟,马洋博,等.第四代制冷剂HFO-1234yf[J].化工新型材料,2010,38(8):30-32,40.
[4]杨刚,杨会娥,李惠黎,等.新型制冷剂——HFO-1234ze和HFO-1234yf[J].有机氟工业,2009(3):16-20,35.
[5]Akasaka R,Tanaka K,Higashi Y.Thermodynamic property modeling for 2,3,3,3-tetrafluoropropene (HFO-1234yf)[J].International Journal of Refrigeration,2010,33(1):52-60.
[6]Mortada S,Zoughaib A,Arzano-Daurelle C,et al.Boiling heat transfer and pressure drop of R-134a and R-1234yf in minichannels for low mass fluxes[J].International Journal of Refrigeration,2012,35(4):962-973.
[7]Akram M W,Polychronopoulou K,Polycarpou A A.Lubricity of environmentally friendly HFO-1234yf refrigerant[J].Tribology International,2013(57):92-100.
[8]Brown J S,Zilio C,Cavallini A.Thermodynamic properties of eight fluorinated olefins[J].International Journal of Refrigeration,2010,33(2):235-241.
[9]Mahler B A,Nappa M J, Knapp J P.Compositions comprising 1,1,1,2,3-pentafluoropropane or 2,3,3,3-tetrafluoropropene:US,20090278075[P].2009-10-12.
[10]Rao V N M,Nappa M J,Sievert A C.Tetrafluoropropene production process:US,20090264689[P].2009-10-22.
[11]Harmon J.Preparation of vinyl fluoride:US,2599631[P].1952-06-10.
[12]Sharratt A P,Low R E,Mccarthy J C.Process for the preparation of c3-7 fluoroalkenes by base-mediated dehydrohalogenatation of hydrohalogenated c3-7 fluoroalkanes:WO,2008075017[P].2008-06-26.
[13]Yousef E M,David F P.Preparation of 1,1,1,3-tetrafluoropropene(1234ze):EP,0974571[P].2000-01-26.
[14]Tung H S,Johnson R C,Merkel D C.Process for the manufacture of 1,1,1,3-tetrafluoropropene:US,20050020862[P].2005-01-27.
[15]Cottrell S A,Tung H S,Wang H.Manufacturing process for HFO-1234ze:US,20100022809[P].2010-01-28.
[16]Sievert A C,Carlson N A.Preparation of hydrofluoroolefins by dehydrofluorination:US,20100174123[P].2010-07-08.
[17]Takahshi K,Chaki T,Shiotani Y.Process for preparing 2,3,3,3-tetrafluoropropene:WO,2010021406[P].2010-02-25.
[18]Nappa M J,Rao V N M.Process for the manufacture of 1,1,1,2,3-pentafluoropropane:US,5396000[P].1995-03-07.
[19]Tavoularis G,Keane M A.Gas phase catalytic dehydrochlorination and hydrodechlorination of aliphatic and aromatic systems[J].Journalof Molecular Catalysis A:Chemical,1999,142(2):187-199.
[20]Gandler J R,Yokoyama T.The E2 transition state:Elimination reaction of 2-(2,4-dinitrophenyl)ethyl halides[J].Journal of the American Chemical Society,1984,106(1):130-135.
[21]Thibblin A.Borderline between E1cB and E2 mechanisms.Elimination of HCl from fluorene derivatives[J].Journal of the American Chemical Society,1988,110(14):4582-4586.
[22]Kamiguchi S,Watanabe M,Kondo K,et al.Catalytic dehydrohalogenation of alkyl halides by Nb,Mo,Ta,and W halide clusters with an octahedral metal framework and by a Re chloride cluster with a triangular metal framework[J].Journal of Molercular Catalysis A:Chemical,2003,203(1-2):153-163.
[23]Teinz K,Wuttke S,Börno F,et al.Highly selective metal fluoride catalysts for the dehydrohalogenation of 3-chloro-1,1,1,3-tetrafluorobutane[J].Journal of Catalysis,2011,282(1):175-182.
[24]Li G L,Nishiguchi H,Ishihara T,et al.Catalytic dehydrofluorination of CF3CH3(HFC143a) into CF2CH2(HFC1132a)[J].Applied Catalysis B:Environmental,1998,16(4):309-317.
[25]Mukhopadhyay S,Tung H S,Puy M V D,et al.Method for producing fluorinated organic compounds:US,20070197841[P].2007-08-23.
[26]Miller R N,Nappa M J,Rao V N M,et al.Azeotrope compositions comprising 2,3,3,3-tetrafluoropropene and hydrogen fluoride and uses thereof:US,7476771[P].2009-01-13.
[27]Miller R N,Nappa M J,Rao V N M,et al.Azeotrope compositions comprising E-1,3,3,3-tetrafluoropropene and hydrogen fluoride and uses thereof:US,7423188[P].2008-09-09.
[28]Mukhopadhyay S,Nair H K,Tung H S,et al.Method for producing fluorinated organic compounds:US,20070129580[P].2007-06-07.
[29]Sakyu F,Hibino Y.Method for producing 1,3,3,3-tetrafluoropropene:US,20090099395[P].2009-04-16.
[30]Puy M V D,Cook G R,Scheidle P H,et al.Process for the manufacture of fluorinated olefins:US,20070179324[P].2007-08-02.
[31]Mukhopadhyay S,Bortz C L,Light B A,et al.Process for synthesis of fluorinated olefins:US,20090099396[P].2009-04-16.
[32]Elsheikh M Y,Bonnet P,Keeley O C N,et al.Dehydrofluorination of pentafluoroalkanes to form tetrafluoroolefins:WO,2011140013[P].2011-10-11.
[33]Hirokazu A,Eiji S.Method for manufacturing 1,1,1,2,3-pentafluoropropene and method for manufacturing 1,1,1,2,3-pentafluoropropane:EP,0726243[P].1996-08-14.
[34]Nappa M J,Jackson A.Catalysts and process to manufacture 2,3,3,3-tetrafluoropropene:US,20120172639[P].2012-07-05.
[35]Christoph F J,Coulston G W,Rao V N M.Treatment of chromium oxide and catalytic manufacture of vinyl fluoride:WO,9641679[P].1996-12-27.
[36]Sievert A C.Method for producing 1,1,1,2,3-pentafluoropropene and related azeotropic compositions:WO,2008030444[P].2008-03-13.
[37]Nappa M J,Rao V N M.Catalytic production processes for making tetrafluoropropenes and pentafluoropropenes:US,20100004492[P].2010-01-07.
[38]Rao V N M,Munirpallam A S.Fluoroolefin manufacturing process:US,6031141[P].2000-02-29.
[39]Rao V N M,Sievert A C,Nappa M J.Process to manufacture 2,3,3,3-tetrafluoropropene:WO,2008030440[P].2008-03-13.
[40]Powell R L,Sharratt A P.Process for the production of fluorine containing olefins:WO,9605157[P].1996-02-22.
[41]Smith J W,Mcguiness C,Sharratt A P,et al.Process for the preparation of 2,3,3,3-tetrafluoropropene:WO,2009138764[P].2009-11-19.
[42]Amos T G,Rao V N M,Sievert A C,et al.Chromium oxide compositions containing zinc,their preparation,and their use as catalysts and catalyst precursors:WO,2005037431[P].2005-04-28.
[43]Miller R N,Nappa M J,Rao V N M,et al.Process for production and purification of hydrofluoroolefins:US,20060106263[P].2006-05-18.
[44]Mahler B A,Nappa M J,Knapp J P.Compositions comprising 1,1,1,2,3-pentafluoropropane or 2,3,3,3-tetrafluoropropene:WO,2009137656[P].2009-11-12.
[45]Uenveren E,Kemnitz E,RÜdiger S,et al.Preparation of halogen and hydrogen containing alkenes over metal fluoride catalysts:WO,2009010472[P].2009-01-22.
[46]Nappa M J,Rao V N M.The catalytic manufacture of vinyl fluoride:WO,9822414[P].1998-05-28.
[47]Merkel D C,Pokrovski K A,Tung H S.Methods to produce 3,3,3-trifluoropropene:US,20120059201[P].2012-03-08.
[48]Elsheikh M Y,Bonnet P,Wendlinger L.Two step process for the manufacture of hydrofluoroolefins:WO,2009003157[P].2009-12-31.
[49]Sheppard B M,Yousef E M.Dehydrofluorination and dehydrogenation of fluorinated alkenes:EP,0406748[P].1991-1-09.
[50]Sievert A C,Nappa M J,Rao V N M.Process for the manufacture of 1,1,1,3,3-pentafluoropropane:WO,9833756[P].1998-08-06.
[51]Nappa M J,Rao V N M.The catalytic manufacture of pentafluoropropenes:WO,9833755[P].1998-08-06.
猜你喜欢
化工管理(2022年13期)2022-12-02
中国特种设备安全(2018年12期)2018-03-15
能源(2017年5期)2017-07-06
中国卫生标准管理(2015年17期)2016-01-20
西北工业大学学报(2015年4期)2016-01-19
中国当代医药(2015年9期)2015-03-01
中国塑料(2014年12期)2014-10-17
应用技术学报(2014年1期)2014-02-28
河北工业科技(2014年3期)2014-02-27
无机盐工业(2013年1期)2013-03-19
