
高碱煤中NaCl与水冷壁吸附作用的量子化学研究
2013-10-10 12:10杨换凌张忠孝乌晓江
上海理工大学学报 2013年5期
杨换凌, 张忠孝, 乌晓江
(1.上海理工大学 能源与动力工程学院,上海 200093;2.上海锅炉厂有限公司 技术研发室,上海 200245)
新疆准东煤是我国近年已探明的最大煤田,煤炭资源预测储量3 900亿吨,它将成为我国又一重要的能源与电力建设基地[1].由于准东煤中的原生矿物富含大量碱金属和碱土金属,特别是煤中的Na含量远高于现有已知动力用煤中Na含量(煤灰中Na2O含量一般在4%~10%之间,而常规动力用煤则在2%以下),致使目前现役燃煤锅炉在燃用准东煤时均出现严重的炉膛结渣和高温受热面(高过、高再)沾污、堵塞等问题.煤燃烧过程中,煤中矿物质不仅发生复杂的物理-化学变化,而且高温下煤中Na,K等碱金属还会发生升华、冷凝现象,在烟气中形成气态碱金属或气态碱金属氧化物,在冷凝、团聚的作用下亚微米级飞灰颗粒通过热泳沉积或化学反应等途径粘结到受热面表面,形成沾污初始层[2].前人研究表明:受热面表面初始层对灰的沾污起着至关重要的作用[3],这不仅与颗粒的物理化学性质有关,还与受热面表面物质化学成份、表面光洁度等有关,受热面表面亚米级颗粒熔融过程中表面张力的大小直接反映其沾污强度的大小,而物质相变过程中表面张力的变化特性取决于物质本身的物理化学特性[4].
随着量子理论和量子化学计算方法的发展与进步,应用量子化学理论从物质分子结构的物理-化学特性角度深入揭示物质的宏观物理-化学特性,从而为分析物质在相变过程中的物理-化学特性提供理论依据.张忠孝对煤灰中的钙长石、钠长石、铁橄榄石、莫来石等耐熔矿物和低熔融矿物进行量子化学计算研究,探索高温条件下煤灰熔融过程中矿物 质 间 的 复 杂 物 理 化 学 反 应 机 理[5-7];von Oertzen[8]利用量子化学Mulliken布居分析黄铁矿(100)面和黄铁矿电子结构之间的差异;Yu[9]研究了Na在Cu(111)面的吸附;Trubitsyn[10]通过对氯化镁和一氧化碳的吸附研究,发现活化的氯化镁的吸附位点与实验数据吻合得很好;谭晓朝[11]利用第一性原理研究氧吸附在铁(110)面;郭欣等[12]利用密度泛函理论研究分子在簇模型CaO(001)面上吸附;张蓉等[13]利用密度泛函理论研究Cl-在Al(100)表面的吸附;肖海燕等[14]利用密度泛函理论研究Na,K在Rh(111)面的吸附;任华[15]利用双层界面模型模拟大分子之间的吸附.这些研究是量子理论在晶体表面性质、分子在晶体表面的吸附、离子在晶体表面的吸附等模拟计算上的应用,而从晶体表面与晶体表面之间的吸附作用的研究目前还没有具体的文献支持,因此本文应用量子理论模拟晶体表面与表面之间的吸附作用,从微观角度解释锅炉结渣机理,这属于一个探索性计算.
1 计算方法和模型
1.1 计算方法
本文采用了基于第一性原理(ab initio)的超软赝势(ultrasoft pseudo-potential)[16]平面波法,结构优化采用BFGS算法[17],利用 Materials Studio软件工具栏中的Build菜单对晶体进行切割和界面建模,采用了广义梯度近似(GGA)中的PBE算法描述电子交换相关作用,结构优化及能量计算采用Materials Studio软件包中的Castep模块.为了保证计算精度,总能量精度为1.0×10-5eV·atom-1,能量截断值为300.0eV,每个结构单元应力小于0.05GPa,每个原子所受的晶体内作用力小于0.03eV·nm-1,布里渊区K点取样为2×3×1.结构模型采用BFGS算法进行优化与弛豫.周期性边界条件选取这些物质的晶体原胞.
1.2 计算模型
Castep几何优化是基于减小计算力和应力的数量级,得到最优化晶格常数.锅炉炉内受热面材质一般为钢,改变受热面材质常做Al和Ni涂层.因此,本文选取的3种计算材料分别为Fe,Al,Ni.但是由于锅炉受热面在工作中一般会被氧化,所以本文选用Fe2O3,Al2O3,NiO为研究对象,对应的晶体参数如表1所示,a,b,c分别指晶胞的边长.图1为经过几何优化后得到的4种物质三维结构示意图.
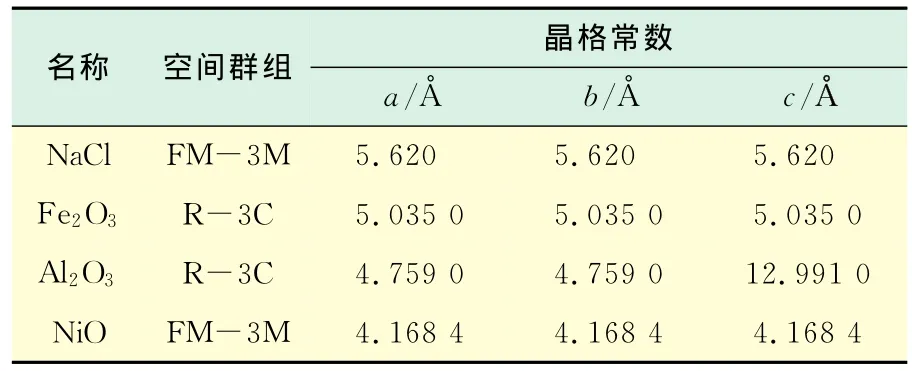
表1 NaCl,Fe2O3,Al2O3,NiO晶体参数Tab.1 Lattice constants of NaCl,Fe2O3,Al2O3,NiO
1.2.1 单层晶面的建立

图1 晶体的3D结构Fig.1 3Dstructure of the crystal
要研究两种物质之间的吸附,一般从面与面之间作用力着手.NaCl及Fe2O3,Al2O3,NiO物质在常温下无解理面(矿物晶体在外力作用下严格沿着一定结晶方向破裂,并且能裂出光滑平面的性质称为解理,这些平面称为解理面),所以在晶体建模前利用Materials Studio工具栏中的Build对优化后的3D结构模型进行处理.一般金属氧化物的(110)面比较稳定,易于进行界面建模,因此对NaCl,Fe2O3,Al2O3及NiO优化后的3D结构模型进行(110)面切割.表面的深度越厚,计算的准确性越高.但是考虑到计算时间的长短以及计算机的性能,一般切割5~8层便认为计算有效.在此基础上分别构建出NaCl(110),Fe2O3(110),Al2O3(110),NiO(110)清洁表面,如图2所示.

图2 晶体的(110)网面Fig.2 Net surface of the crystal(110)
1.2.2 双层界面模型的建立
将NaCl(110)分别与Fe2O3(110),Al2O3(110),NiO(110)晶体表面利用 Materials Studio分层建模工具进行建模,所建模型如下页图3所示.若要z方向(晶体切割表面的径向)原子层不相互作用,则必须建立足够厚度的真空层,通常厚度达到10Å以上即能满足要求.同时考虑到建模后整个模型实际上是两个层交替出现的周期排列(…ABABABABAB…),为消除在模拟过程中出现干扰,在上层的上方也加入10Å的真空层.经过界面建模后得到的几何参数如表2所示.
表面在一般条件下只有弛豫,不会发生表面重构,即表面层及其附近的原子只在垂直于表面的方向上下移动.

表2 吸附模型建立后的几何参数Tab.2 Geometrical parameters of the adsorption model

图3 晶体相互作用模型Fig.3 Model of crystal interaction
表2列出了 NaCl(110)面分别与Fe2O3,Al2O3及NiO的(110)面建立界面模型后的几何结构参数.对比表1,界面建模后晶格参数均发生变化,计算结果会出现偏差.这里需要指出,界面建模的过程中,可以通过建立超晶结构的方法使上、下层尺寸达到相近,但由于上述物质为大分子体系,受限于计算时间和计算机内存等方面的影响,同时为了便于数据的采集,上、下层物质均采取适当的近似,这些近似都在误差允许的范围内.因此,本文计算中的误差可以忽略不计.
TYRONE:I thought you'd gone back uptown to meet him.
2 计算结果与讨论
2.1 反应活性的计算
在界面模型几何优化后得到稳定结构的基础上,进行其化学活性的有关计算,吸附模型见图3,模型的上层均为NaCl(110)面,下层分别为Fe2O3(110),Al2O3(110),NiO(110)面.计算得到了原子的静电荷数,如表3所示.
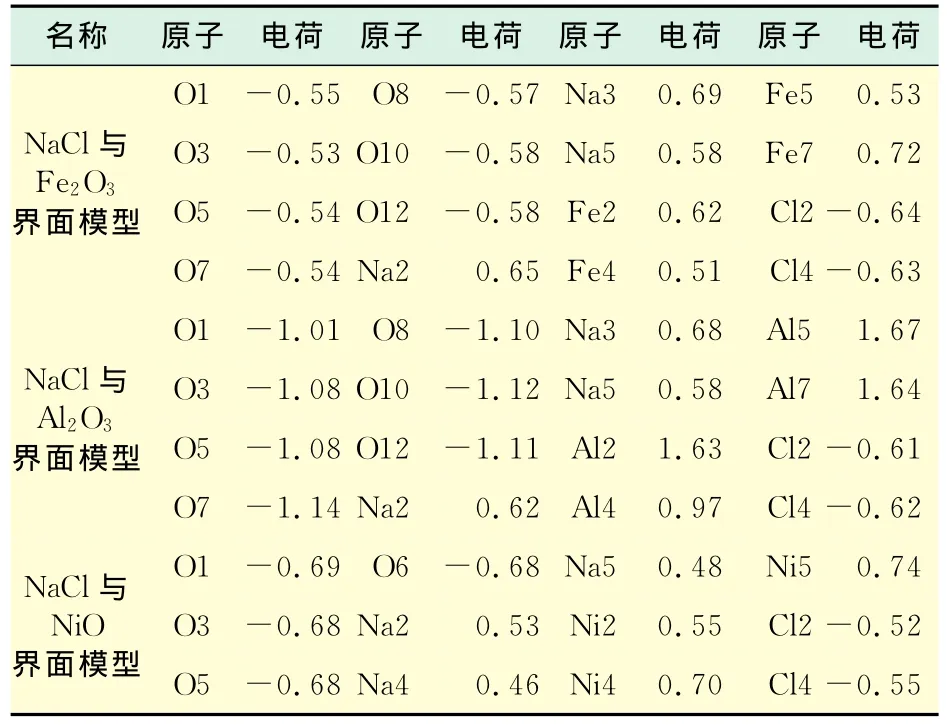
表3 吸附模型的部分原子电荷数Tab.3 Atomic charges of the adsorption model
表3分别列出NaCl与Fe2O3,Al2O3及NiO构成界面模型的部分原子静电荷数,比较原子静电荷数,发现其中O及Cl原子均带负电荷,而Na及Fe,Al,Ni均带正电荷.这是由于吸附导致金属表面电子向被吸附O原子转移使O原子带负电,金属表面带正电,电荷数发生微弱变化.这一现象说明燃煤过程中,挥发性NaCl与锅炉受热面接触发生吸附作用.这与结渣是由于含钠物质与受热面吸附形成初始层的预测相符合.
对比表4中物质洁净的(110)表面与建立界面模型后键长,表面吸附使相应的键长发生微量的变化.NaCl与Fe2O3的吸附模型中,键长普遍被拉长,可见灰粒与受热面接触时,挥发性NaCl与受热面产生稳定的吸附作用,从而使灰粒牢牢地粘结在受热面上,造成锅炉受热面的严重结渣.而NaCl与NiO的吸附模型中,键长既有拉长也有压缩变化,说明吸附过程中部分原子之间产生排斥作用而不能形成稳定的吸附作用,表现为涂Ni受热面上结渣并不明显.
2.2 吸附模型吸附能的计算
当体系能量收敛后,表明体系达到平衡状态,接下来进行吸附能的计算.首先将结构稳定的模型恢复下层固定的分子,然后分别对Et,Es1,Es2进行计算,其吸附能的计算公式[15]为
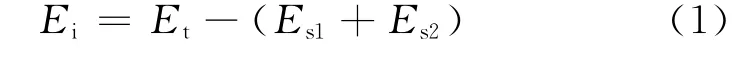
其中,Et定义为吸附后吸附体系的总能量;Es1定义为除去NaCl(110)后表面的能量;Es2定义为除去金属氧化物表面后NaCl(110)表面的能量.吸附能为正,代表吸附为放热反应,且数值越大说明相互作用越强,吸附过程越容易发生,吸附结构越稳定.通过计算得出以下数据,如表5所示.
利用式(1)得出
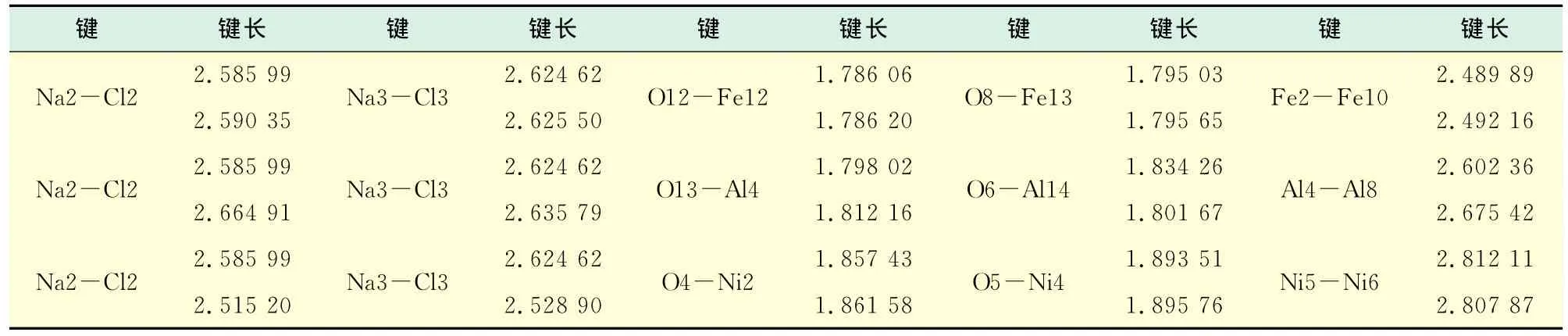
表4 优化后洁净面及界面模型部分键长数据Tab.4 Part of the bond length data of the optimized clean surface and interface model

表5 界面模型表面的能量Tab.5 Energy onthesurfaceofinterfacemodel
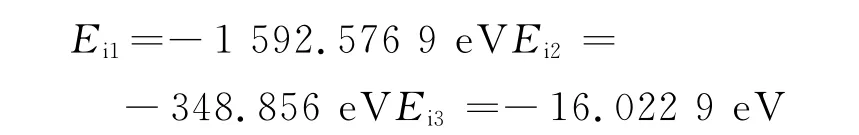
考虑到构建的模型中所包含的原子数不同,吸附能力可能与单原子的吸附能相关,因此在表面吸附能的基础上计算原子平均能Eb,原子平均能量定义为[18]

其中,N为表面吸附模型的原子个数,结合表2数据利用式(2)得出
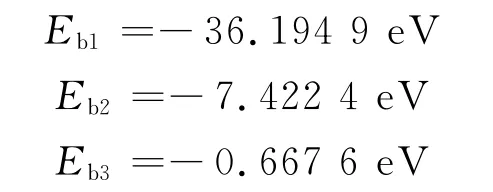
以上计算吸附模型为图3,经过模拟计算得出结果:NaCl与Fe2O3的吸附能为-1 592.576 9eV,数值最大;NaCl与Al2O3吸附能为-348.856eV;NaCl与NiO之间的吸附能为-16.022 9eV,几乎可以忽略,而转换为原子平均能后这一大小关系不变.计算结果表明:钢制材料的锅炉燃煤过程中灰粒与受热面的相互作用强;作涂Al处理的锅炉燃煤过程中灰粒与受热面次之;作涂Ni处理的锅炉燃煤过程中灰粒与受热面最弱.这说明,与灰粒相互作用强的钢制受热面产生了较强的吸附作用,能快速且大量形成初始沉积层;而与灰粒相互作用弱的涂Ni受热面吸附作用极小,受热面表面没有明显的积灰结渣现象.这个结果为利用吸附能讨论结渣特性提供数据支持.
2.3 结渣机理的探索
固体表面上的原子或分子的力场和液体表面一样也是不均衡的,因此,也有自发降低表面能的倾向.由于固体表面不像液体表面而难于收缩,所以只能靠降低界面张力的办法来降低表面能,因此在固体表面也会产生吸附作用[19].
燃煤锅炉炉内结渣是一个复杂的物理化学过程,如图4所示,燃煤锅炉中飞灰颗粒以及灰渣中的某些成分选择性沉积或熔融性沉积形成初始层[20-21].沾污层热阻很大,灰层表面的温度不断提高,会使积灰结渣过程迅速增长,碱金属Na在初始沉积层形成过程中起着助熔剂和增加对管壁的附着能力的作用[22].通过对 NaCl与Fe2O3,Al2O3及 NiO之间的活性剂吸附能的模拟计算,可以看出NaCl与Fe2O3之间的吸附能力高,使之容易粘结在受热面形成严重的结渣;作涂Al2O3处理的受热面吸附能力较低,它们之间没有形成稳固的吸附模型;涂Ni处理的受热面吸附能力最弱,几乎不吸附.通过实验室试烧新疆准东煤实验,实验结果表明作涂Al处理的水冷壁结渣情况有所好转,而作涂Ni处理的水冷壁几乎不结渣,这些计算结果与实验结果相吻合.
3 结 论
a.高碱煤燃烧造成锅炉结渣沾污是一个十分复杂的物理化学问题,通过计算挥发性物质Na与受热面之间的吸附作用能,为寻找解决锅炉结渣提供了有效途径.
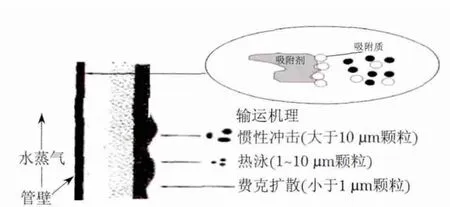
图4 灰渣沉积过程示意图Fig.4 Diagram of ash deposition process
b.高碱煤中富含Na和K等物质及其化合物,高温下易于挥发、升华,与受热面接触产生稳定吸附,形成初始沉积层.由于沾污层热阻很大,会使灰层表面的温度不断提高,造成局部温度过高,加剧炉内沾污结渣现象.
c.利用Materials Studio软件包中分层建模工具建立双层界面模型,利用Castep模块并结合GGA进行优化及计算,得出Fe2O3(110)与NaCl(110)的吸附能为-1 592.576 9eV,Al2O3(110)与NaCl(110)的吸附能为-348.856eV,NiO(110)与NaCl(110)的吸附能为-16.022 9eV.根据相互作用能的大小,对比来自电厂方面结渣数据,吸附能越大,结渣情况越严重,吸附能越小,结渣情况越轻,说明吸附能的大小与结渣的强弱相关.
d.通过量子化学计算结渣初始层中主要矿物质NaCl与不同表面材质(Fe,Al,Ni)的受热面吸附能,可以预测结渣趋势,从而可以通过管壁涂层等方法改善炉内结渣状况.
[1]杨忠灿,刘家利,何红光.新疆准东煤特性研究及其锅炉选型[J].热力发电,2010,39(8):38-44.
[2]Benson S A,Steadman E N,Zygarlicke C J,et al.Application of advanced technology to ash-related problems in boilers[M].NewYork:Plenum Press,1996.
[3]顾志恩,柳美瑛.锅炉结渣机理及煤潜在结渣特性[J].电力勘测设计,2006(2):39-42.
[4]何佩教,张忠孝.我国动力用煤结渣特性的试验研究[J].动力工程,1987(2):1-11.
[5]李洁,杜梅芳,闫博,等.添加硼砂助熔剂煤灰熔融性的量子化学与实验研究[J].燃料化学学报,2008,36(5):519-523.
[6]陈玉爽,张忠孝,乌晓江,等.配煤对煤灰熔融性影响的实验与量化研究[J].燃料化学学报,2009,37(5):521-526.
[7]管嵘清,杜梅芳,李洁,等.高熔点煤灰添加硼砂助熔的理论和实验研究[J].上海理工大学学报,2009,31(4):362-366.
[8]von Oertzen G U,Skinner W M,Nesbitt H W.Ab initio and XPS studies of pyrite (100)surface states[J].Radiation Physics and Chemistry,2006,75 (11):1855-1860.
[9]Yu S,Bahrim B,Makarenko B,et al.Local effect of Na adsorption on Cu(111)[J].Surface Science,2012,606(21/22):1700-1704.
[10]Trubitsyn D A,Zakharov V A,Zakharov I I.A theoretical investigation of the adsorption surface sites of the activated MgCl2[J].Journal of Molecular Catalysis A:Chemical,2007,270(1/2):164-170.
[11]Tan X C,Zhou J C,Peng Y Q.First-principles study of oxygen adsorption on Fe(110)surface[J].Applied Surface Science,2012,258 (22):8484-8491.
[12]郭欣,郑楚光,吕乃霞,等.簇模型CaO(001)面上吸附汞与氯化汞的密度泛函理论研究[J].中国电机工程学报,2005,25(13):101-105.
[13]张蓉,黄开有,张攀,等.Cl-在Al(100)表面吸附的密度泛函理论研究[J].粉末冶金材料科学与工程,2010,15(5):434-438.
[14]Xiao H Y,Xie D Q.Density functional study of the adsorption of Na and K on Rh(111)[J].Surface Science,2004,553(l/2/3):13-22.
[15]任华.分子模拟在界面相互作用计算中的应用[D].西安:西北工业大学,2007.
[16]Vanderbilt D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Phys Rev B,1990,41(11):7892-7895.
[17]Fischer T H,Almlof J A.General methods for geometry and wave function optimization [J].J Phys Chem,1992,96(24):9768-9774.
[18]Neugebauer J,Scheffler M,Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111)[J].Phys Rev B,1992,46(24):16067-16080.
[19]赵振国.吸附作用原理[M].北京:化学工业出版社,2005.
[20]池作和,周昊,蒋啸,等.锅炉结渣机理及防结渣技术措施研究[J].热力发电,1999(4):26-30.
[21]马其良,周世平,顾士龙,等.300MW机组炉内结渣原因分析及防止[J].上海理工大学学报,2001,23(2):36-41.
[22]盛昌栋,张军,徐益谦.煤中含铁矿物在煤粉燃烧过程中行为的研究进展[J].煤炭转化,1998,21(3):14-18.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
人工晶体学报(2021年10期)2021-11-26
发电设备(2019年3期)2019-08-13
合成材料老化与应用(2015年1期)2015-03-23
黑龙江电力(2014年6期)2014-03-06
中国新技术新产品(2012年21期)2012-12-28
