
基于融合核方程对药物-靶点作用预测研究
2013-09-26 03:46郝理阳
电子设计工程 2013年23期
郝理阳,潘 泉
(西北工业大学 自动化学院,陕西 西安710072)
药物-靶点作用研究是药物研发的关键[1]。药物研发周期长,花费高[2]。通过计算方法研究药物-靶点作用能够有效缩小实验范围,降低成本[3]。自2001年第一张人类基因组图谱草图绘制完成,人类已进入后基因时代,随着测序技术日益成熟,基因序列、蛋白质序列等相关数据进入指数增长阶段[4]。面对海量数据,借助计算方法分析、挖据其中有用信息研究药物-靶点作用,对于开发具有高选择性的药物有着重要意义[5]。
药物-靶点作用的传统研究方法分为配体方法和结构方法。配体方法以靶点作用结合位点信息为基础,建立生物靶点模型,构造与之作用的药物分子[6]。结构方法以靶点蛋白质三维结构为基础[7],通过现有数据库构建药物分子。然而,已知配体信息或者蛋白质三维结构的蛋白质数量有限,从而限制了传统方法在大规模数据上的应用[8-9]。
机器学习方法是一类人工智能方法,常被应用于数据挖掘,计算机视觉和信号处理等领域。近几年,机器学习方法被引入药物-靶点作用研究[10]。药物-靶点作用预测问题可以看成一种广泛意义上的链路预测问题[11],即利用已知网络节点以及网络结构等信息预测网络中尚未产生连接的两个节点之间产生连接的可能性[12]。基于该思想的几种模型包括:最近邻模型、二分图学习模型、二分局部模型、高斯相互作用核模型。其中,Van-Laarhoven等人在2011年提出的高斯作用核模型所得到的结果最优[10]。该模型通过计算作用向量之间的欧氏距离、药物分子结构相似性、蛋白质序列相似性,利用正则化最小二乘分类器对药物-靶点作用进行预测。然而该模型并未引入表征药物作用特征的物化信息。本文提出基于融合核方程预测药物-靶点作用 (Prediction of Drug-target Interactions by Integrated Kernel Function, DTIKF)算法,该算法从药物分子物化信息、蛋白质序列信息以及药物-靶点作用信息3个方面提取药物-靶点特征向量,利用融合的核方程将药物分子、蛋白质分子这两个异相物质映射到同一个空间内,利用支持向量机(Suppor Vector Machine,SVM)思想对药物-靶点进行预测,实验结果显示该算法不仅可以有效预测已知药物-靶点之间的潜在作用,并可以预测新药物与蛋白质之间的相互作用情况。
1 数据库与方法
1.1 数据库
Drugbank 数据库(http://www.drugbank.ca)3.0 版本[13]收录了 种药物、 种靶点。其中包括 种美国食品药品监督管理局批准的小分子药物,131种蛋白质和多肽类生物技术药物,85种营养制品,5 080种处于实验研究阶段的药物。每一个药物卡片都包含了与该药物相关的约150类信息,包括药物的化学分子式、分子重量、化学结构、剂型、药物类型、药物种类、疏水性、溶解度、半衰期、分子状态、各类数据库的索引名、文献等化学、药理学、医学方面的信息,每一个药物对应的靶点信息包括该靶点的蛋白质序列、基因序列、分子重量、残基数目、通路、细胞位置、各类数据库索引名、文献等信息。从中去除药物分子量小于100的药物样本,最后得到1 516个靶点,3 712个药物,以及它们之间7 719个药物-靶点作用对,这7 719条作用对为本文实验中药物-靶点作用数据集的正集。
将这1 516个靶点与3 712个药物之间两两组合,除去已知的药物-靶点作用对,剩余的药物-靶点组合对即为没有作用的数据集合,即负样本集。例如,对于药物和靶点,之间存在相互作用对和,则将和看作是没有相互作用的样本。可以很容易看出,没有作用的药物-靶点对的数量远远大于有作用的药物-靶点对的数量,为个,从中随机挑选7 719个无作用的药物-靶点样本,构成本文实验中药物-靶点作用数据集的负集。
1.2 DTIKF-SVM预测方法
1.2.1 药物-靶点二元模型建立
由药物-靶点作用信息构建药物-靶点作用二元网络。该二元网络可用关联矩阵 Φ 描述,Φ=(φij)m×n,m=3 712,n=1 516:

其中,若药物与靶点有作用,那么φij=1,反之φij=0。这样,Φ中的每一行表征一种药物的作用信息,Φ中的每一列表征一个靶点的作用信息,定义药物di的作用信息向量xdi和靶点ti的作用信息向量yij如下:

本文旨在通过已知的作用信息向量xdi和ytj,预测新药物、靶点的作用信息。
1.2.2 药物-靶点作用信息
对于每一个药物di,其作用信息向量xdi表示该药物与所有靶点tj作用情况;同样,对于每一个靶点,其作用信息向量ytj表示该靶点与二元网络中所有药物作用的信息。利用径向基方程的形式,构建药物作用信息函数k1d:

同样,建立靶点作用信息函数k1t:

药物、靶点作用信息函数分别度量二元网络中药物之间、靶点之间距离的远近程度。其中, 为径向基方程的带宽,h越小,径向基方程的宽度就越小,选择性就越高。药物、靶点作用信息函数带宽hd、ht分别定义为:
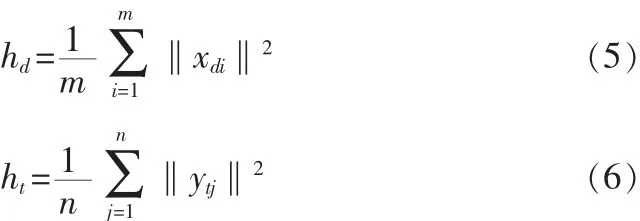
通过参数hd、ht,药物、靶点作用信息函数被归一化,为下一步融合药物分子物化信息和蛋白质序列信息做好准备。
1.2.3 融合药物分子物化信息和蛋白质序列信息
根据文献[7],有4种原子类型对药物发生作用与否的影响最大,分别是疏水原子,氢键供体,氢键受体,极性原子。本文根据这4类原子特性定义药物分子物化特征向量udi:

其中 λ1为分子重量,λ2为疏水性,λ3~λ8分别为碳、 氢、氧、氮、磷、硫六种原子在药物在分子中的概率f与它们各自原子重量q的乘积,即:

蛋白质的序列信息由Smith-Waterman序列相似性比对算法[14]得到,用序列相似矩阵 R=(rij)n×n,n=1 516 表示,R 中的元素rij表示蛋白质i和蛋白质j之间的相似度。易知R为对称矩阵,用ri表示R的第i行。根据药物分子信息和蛋白质序列相似度信息构造药物分子信息函数k2d和蛋白质序列信息函数k2t:

药物分子信息函数、蛋白质序列信息函数分别度量了药物分子之间物化特性的相似程度,以及蛋白质分子之间序列相似程度。根据已有研究结果[1],相似药物更有可能跟同一类靶点反应,而相似靶点更容易与同一类药物进行反应,为了直接反应药物-靶点作用对的关系,将公式(3)、(4)、(9)、(10)融合到一个核方程中,

其中,εd,μt均为 0~1 之间的常数,实验中取 εd=0.5,μt=0.5。
1.3 算法评估参数
本文采用敏感性(Sn)、特异性(Sp)、阳性预测值(PPV)和预测总精度(Q)评估分类系统预测性能。4个参数定义如下:

其中, 表示正确预测的药物 靶点作用对数、 表示正确预测的非作用对数、FP表示错误预测的作用对数、FN表示错误预测的非作用对数。其中,敏感性反映了分类系统对于预测药物-靶点作用对的成功率,特异性反映了确定非作用对的可信度,阳性预测值反映了预测作用对的准确度,预测总精度综合反映了系统预测的准确性。对于一个实用预测系统,既要求有较高的敏感性,也要求有较高的特异性。
2 结果与讨论
SVM的主要思想是建立一个最优决策超平面,使得该平面两侧距平面最近的两类样本之间的距离最大化,从而为分类问题提供良好的泛化能力。利用libsvm3.12[15],更改程序中的核函数为公式(11)的形式,对数据集进行预测。本文采用10 CV(10-fold cross-validation)交叉验证法预测药物-靶点作用。将样本集随机分为不相交的10个子集,然后依次取出一个子集作为测试集,其余的9个子集作为训练集,此过程循环10次。
为验证本文特征提取方法的有效性,分别采用本文特征提取方法与Van-Laarhoven的特征提取方法,基于Van-Laarhoven核方程的支持向量机进行5次10 CV验证试验,实验结果如表1所示。
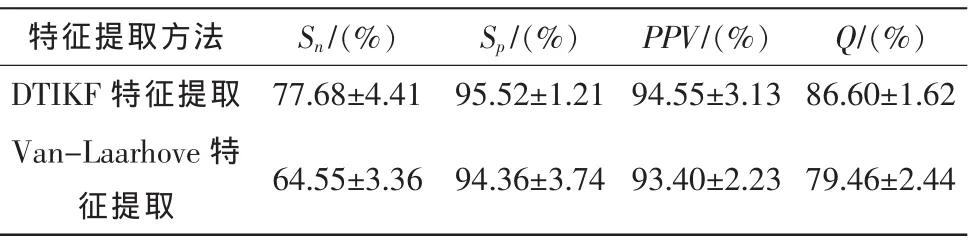
表1 DTIKF、Van-Laarhoven特征提取方法结果Tab.1 Results of DTIKF&Van-Laarhoven feature extraction method
由表1可以看出,整合药物分子物化特性、蛋白质序列相似性以及药物-靶点作用信息的特征提取方法结果优于Van-Laarhoven的方法,总精度为86.60%,提高了7.14个百分点。说明本文特征提取方法能够有效提取药物、靶点作用信息,预测潜在药物-靶点作用。
其次,为了验证本文融合核方程对于药物-靶点作用分类的有效性,采用本文特征提取方法,分别使用基于本文DTIKF核方程的支持向量机,与基于Van-Laarhoven核方程的支持向量机进行5次10CV交叉验证,实验结果见表2。

表2 DTIKF、Van-Laarhoven核方程结果Tab.2 Results of DTIKF&Van-Laarhoven kernel function
由表2可以看出,基于本文核方程的SVM分类器的预测总精度优于Van-Laarhoven的方法。其中,敏感度提高了9.04%,说明在同样的特征提取方法下,DTIKF核对于发现药物-靶点作用的成功率相对于Van-Laarhoven的方法有所提高;特异性提高了4.28%,说明基于融合核方程的SVM分类器在确定非药物 靶点作用的可信度有所提高,在实际应用预测系统中,将使所得结果具有较高置信度;阳性预测值提高了5.21%,表明DTIKF-SVM预测药物-靶点作用对的准确度有所提高,在实际应用中,较高的阳性预测值能够有效缩小实验范围,提高实验效率,降低实验成本;预测总精度比Van-Laarhoven的方法提高了6.65%,说明了本文融合核对于预测药物-靶点作用的有效性。
由表1、表2可以看出,整合药物分子物化信息、蛋白质序列相似性以及药物-靶点作用信息的特征提取方法能够有效挖掘药物、靶点作用对的特征,提高分类器的性能;基于融合核的支持向量机能够更加有效的预测药物-靶点作用对,提高分类性能。
3 结 论
文中通过融合药物分子物化信息,蛋白质序列信息和药物-靶点作用信息,利用基于支持向量机的分类系统对药物-靶点作用进行预测。并通过敏感性、特异性、阳性预测值以及总体精度对本文提出的融合算法和Van-Laarhoven的算法进行对比。实验结果显示,本文的特征提取方法能够有效挖掘药物-靶点作用对的生物学信息,提高预测精度;融合核方程将药物-靶点作用信息与生物化学信息有效融合,提高了支持向量机在分类预测药物-靶点作用的精度。预测的潜在药物-靶点作用仍需要实验方法验证,但本文提出的计算方法可作为实验方法的一种辅助,降低实验成本,缩短药物研发周期,在实际应用中具有重要价值。
[1]Yamanishi Y,Araki M,Gutteridge A,et al.Prediction of drug-target interaction networks from the integration of chemical and genomic spaces[J].Bioinformatics,2008,24(13):232-240.
[2]DiMasi J A,Hansen R W,Grabowski H G.The price of innovation:new estimates of drug development costs[J].Journal of Health Economics,2003,22(2):151-186.
[3]Paul S M,Mytelka D S,Dunwiddie C T,et al.How to improve R&D productivity:the pharmaceutical industry’s grand challenge[J].Nature Reviews Drug Discovery,2010,9(3):203-214.
[4]张春霆.生物信息学的现状与展望[J].世界科技研究与发展,2000,22(6):17-20.
ZHANG Chun-ting.The Current Status and The Prospect of Bioinformatics[J].World sic-tech R&D,2000,22(6):17-22.
[5]Davit B M,Nwakama P E,Buehler G J,et al.Comparing generic and innovator drugs:a review of 12 years of bioequivalence data from the United States Food and Drug Administration[J].The Annals of pharmacotherapy,2009,43(10):1583-1597.
[6]Güner O F.Pharmacophore perception, development, and use in drug design[M].La Jolla,International University Line,2000:339-362.
[7]Wang R,Gao Y,Lai L.LigBuilder:A Multi-Purpose Program for Structure-Based Drug Design[J].Journal of Molecular Modeling,2000,6(7-8):498-516.
[8]Jacob L,Vert J P.Protein-ligand interaction prediction:an improved chemogenomics approach[J].Bioinformatics,2008,24(19):2149-2156.
[9]Rarey M,Kramer B,Lengauer T,et al.A fast flexible docking method using an incrementalconstruction algorithm[J].Journal of molecular biology,1996,261(3):470-489.
[10]Van-Laarhoven T,NabuursS B,MarchioriE.Gaussian interaction profile kernels for predicting drug-target interaction[J].Bioinformatics,2011,27(21):3036-3043.
[11]Lü L,Zhou T.Link prediction in complex networks:A survey[J].Physica A:Statistical Mechanics and its Applications,2011,390(6):1150-1170.
[12]Liben-Nowell D,Kleinberg J.The link-prediction problem for social networks[J].Journal of the American society for information science and technology,2007,58(7):1019-1031.
[13]Knox C,Law V,Jewison T,etal.DrugBank 3.0:a comprehensive resource for ‘omics’ research on drugs[J].Nucleic acids research,2011,39(suppl 1):D1035-D1041.
[14]Smith T F,Waterman M S.Identification ofCommon Molecular Subsequences[J].Molecular Biology,1981,147:195-197.
[15]Chang C C,Lin C J.LIBSVM:a library for support vector machines[J].ACM Transactions on Intelligent Systems and Technology,2011,2(27):1-27.
猜你喜欢
新高考·高一数学(2022年3期)2022-04-28
中老年保健(2021年3期)2021-12-03
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19
中国生殖健康(2020年7期)2020-12-10
电子制作(2019年15期)2019-08-27
电子制作(2018年19期)2018-11-14
自动化学报(2017年11期)2017-04-04
高中生学习·高三版(2016年9期)2016-05-14
新高考·高二数学(2015年11期)2015-12-23
医学研究杂志(2015年7期)2015-06-22
