
氧化应激与DNA损伤
2013-09-20 03:05冉茂良
动物营养学报 2013年10期
冉茂良 高 环 尹 杰 陈 斌*
(1.湖南农业大学动物科学技术学院,长沙 410128;2.中国科学院亚热带农业生态研究所,中国科学院亚热带农业生态过程重点实验室,湖南省畜禽健康养殖工程技术中心,农业部中南动物营养与饲料科学观测实验站,长沙 410125;3.中国科学院研究生院,北京 100049)
人和动物机体内由于细胞呼吸和能量代谢时刻发生着有氧氧化,在细胞中产生活性氧分子(reactive oxygen species,ROS),如超氧阴离子(·O-2)和羟自由基(·OH)等自由基。当细胞受到内外环境的刺激后,ROS产生增多,从而破坏了机体氧化与抗氧化系统之间的平衡,最终导致氧化应激(图1)[1-3]。过多的ROS能够激活核因子E2相关因子2(Nrf2)、核转录因子 -κB(NF-κB)、丝裂原活化蛋白激酶(MAPKs)等,从而调节许多氧化物质和抗氧化物质相关基因的表达[4-5]。不仅如此,蛋白质和DNA也是ROS的攻击靶标,ROS能够造成蛋白质结构突变或丧失生物活性、DNA链断裂、DNA位点突变、DNA双链畸变和原癌基因与肿瘤抑制基因突变,最终导致机体产生氧化损伤[6-7]。DNA作为生物体最重要的遗传物质能够保持其自身一定的稳定性,但是DNA却经常发生内在的自发性损伤和遭受X射线、紫外线、烷化剂、嵌入剂等外在刺激而发生损伤,据统计每个细胞内的DNA在24 h内出现10 000次损伤。而DNA损伤能够加速细胞的衰老和凋亡、引起癌症和肿瘤等疾病。不仅如此,有研究报道DNA损伤也可以诱导ROS的产生[8],且对细胞死亡和自救有重要的作用[9],其原因可能是由于ROS能调节p53基因(一种抗癌基因)的活性[10-11]。由此可见,ROS可引起DNA损伤,而DNA损伤也可以诱导产生ROS,因而氧化应激和DNA之间存在着一定的联系。
1 ROS和氧化应激信号通道
1.1 ROS的产生
大量研究表明,ROS有2种来源:一种是内源性,线粒体呼吸链氧化磷酸化过程活性氧泄漏、过氧化酶体和被激活的炎性细胞等[12];另一种是外源性,例如外源物、病原体、促炎细胞因子和重金属等[13]。尽管细胞色素 p450氧化酶也能产生ROS,但线粒体仍是细胞ROS最重要的来源。研究表明,在线粒体呼吸链辅酶还原型辅酶Ⅰ(NADH)、泛醌氧化还原酶和泛醌细胞色素C氧化还原酶的作用下,0.15%的氧被转化为ROS,其中主要是·,由于·能攻击细胞内许多大分子(如脂质、蛋白质和DNA)且能通过酶催化反应产生其他 ROS,如·OH和过氧化氢(H2O2)等,因而·被视为“首要 ROS”[15]。·O2-在超氧化物歧化酶(SOD)的催化作用下被转变为H2O2,H2O2通过过氧化氢酶、谷胱甘肽过氧化物酶(GPx)和Fenton反应被清除。其中过氧化氢酶能够通过歧化反应将2分子H2O2转化为1分子O2和2分子H2O,GPx利用一些还原剂将H2O2转化为2分子H2O,Fenton反应则是在Fe2+的催化下将H2O2分解生成·OH[1,16]。尽管·OH的半衰期(大约10-9s)非常短,但其活性很高,几乎对所有大分子(如碳水化合物、核酸、脂质和蛋白质)造成损伤[17](图 2)。

图1 促氧化和抗氧化系统间的平衡模型Fig.1 Balance model between pro-oxidative and anti-oxidative systems[1]
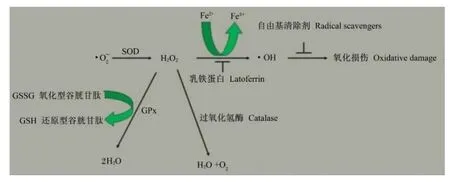
图2 SOD将·转化为H2O2后的已知途径Fig.2 Clear pathways after· transformed into H2O2by SOD
1.2 解耦联蛋白(uncoupling proteins,UCPs)系统的反馈调节
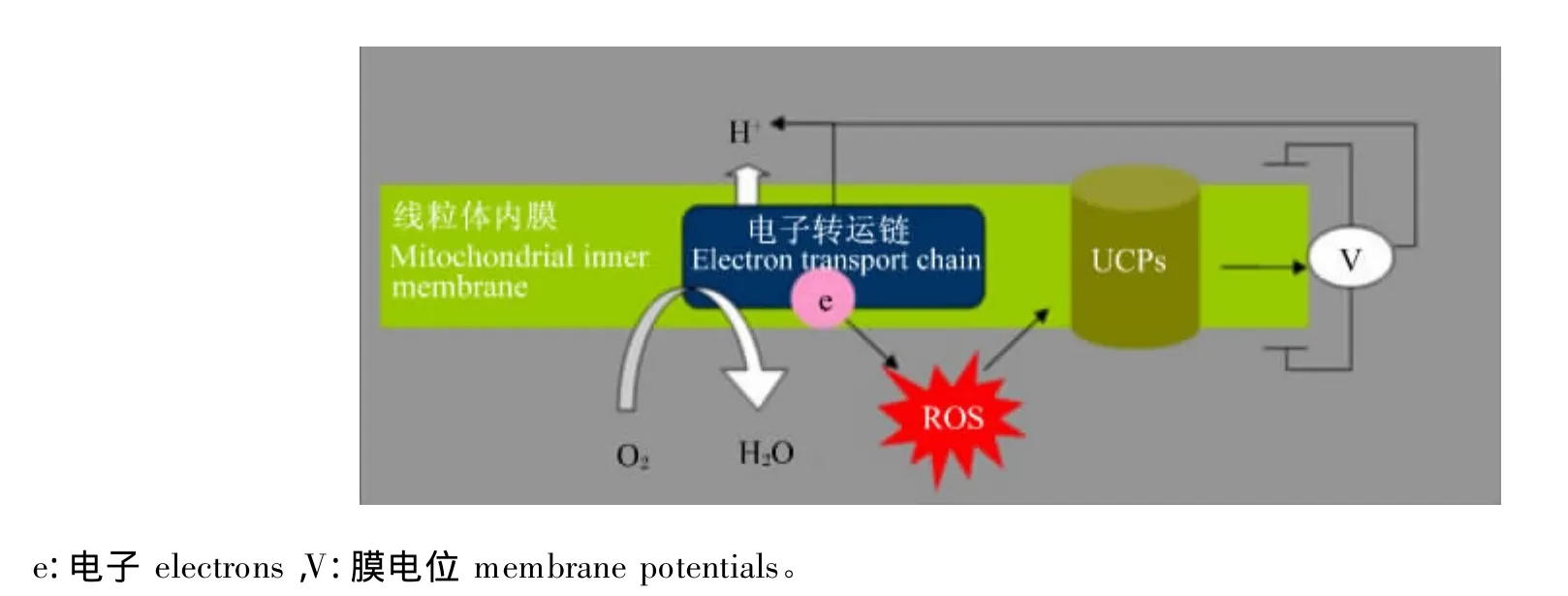
UCPs是线粒体内膜上的一种具有调节质子跨膜作用的特殊蛋白质。目前在哺乳动物体内已经发现 5 种 UCPs,分别为:UCP1、UCP2、UCP3、UCP4 和 UCP5[18-19]。研 究 表 明,在 UCPs 家 族中,仅UCP2和UCP3能够对氧化应激起反馈调节的作用。例如,UCP2基因敲除的小鼠较正常小鼠大脑线粒体在氧化应激下能够产生更多的ROS[20],此外,UCP3 基因的过度表达也能显著降低细胞衰老诱导的ROS的产生[21]。UCPs反馈调节ROS的机制是:UCPs通过降低质子电化学梯度,使呼吸作用中电子传递过程和ATP的合成解耦联,因此将储存的能量以热能的形式释放,并提高静息代谢率,其中UCPs通过调节质子泄漏(proton leak)以降低 ROS 的产量(图3)[22]。研究表明:呼吸链赋予质子的高运动能力,使给氧分子提供1个电子而变成·的电子载体在稳定状态时的浓度升高,由此增加了·的量;·下游衍生物羟基壬烯酸激活电子漏,使膜电位降低,随后通过呼吸链刺激电子流量,使得给氧提供1个电子而变成·的电子载体的在稳定状态时的浓度降低,因此UCPs对ROS的生成有一个负反馈机制,其功能是通过脂质过氧化产物来抵抗氧化应激,但是羟基壬烯酸在UCPs反馈调节ROS 的机制尚不清楚[23-24]。
1.3 氧化应激信号通道
氧化应激能够破坏细胞内氧化还原平衡,从而激活或抑制许多信号通路和一些信号介导分子,如核因子 E2相关因子2-胞质伴侣蛋白(Nrf2/Keap1)信号通道[4]、NF-κB 信号通道[5],MAPKs[25],激酶蛋白 mTOR(一个蛋白质合成的关键调控子)[26]和蛋白激酶 C(PKC)[27]等,最终调节相关基因的表达。其中Nrf2/Keap1是细胞内抵抗氧化应激和保持氧化还原平衡的重要信号通道之一[28],Nrf2是一种氧化应激基本表达的关键转录因子,存在于全身多个器官,它的缺失或激活障碍直接引起细胞对应激源的敏感性变化。因此,本文主要综述了ROS对Nrf2/Keap1信号通路的影响。
Keap1是Nrf2在细胞质中的富含半胱氨酸的结合蛋白,主要通过结合Nrf2使之无法进入细胞核,从而抑制Nrf2的活性,避免引起细胞对应激源的敏感性升高,例如敲除Keap1的基因导致Nrf2信号非常活跃[29-30]。在无应激条件下,Nrf2在细胞质中通过与Keap1结合而被抑制,而当在氧化应激或过量ROS刺激时,半胱氨酸在Keap1中的残留量会增加,随后Keap1作为E3连接酶的活性变弱[31],Nrf2和 Keap1之间的连接被打乱,导致Nrf2泛化和衰退减少,细胞质中自由的Nrf2增多,转移进细胞核的Nrf2增多[32-33],进入细胞核的Nrf2与小Maf蛋白形成异源二聚体,并且和抗氧化反应元件(ARE)连接在一起。随后,ARE被激活并启动抗氧化基因的转录[34],从而使得抗氧化基因得以表达。但是Li等[32]研究报道:细胞内存在2种Nrf2蛋白,一种是游离Nrf2(f Nrf2),另一种是与Keap1结合的Nrf2(kNrf2),在无应激状态下细胞质内绝大多数Nrf2是处于与Keap1结合的状态,只有少量fNrf2进入细胞核以保持氧化还原平衡;在ROS过量时,由于Keap1的自我泛素化使得Keap1与Nrf2的结合量达到饱和或减少[35-36],从而f Nrf2的量增多,进入细胞核强化抗氧化基因的表达[32]。这2种 Nrf2/Keap1信号通道机制的关键不同之处是:前一种认为氧化还原信号是从Keap1到Nrf2的,而后一种认为Keap1和Nrf2都对氧化还原信号有高度的敏感性[32]。但是如前面叙述的细胞内在无应激状态下也时刻都产生ROS,因而细胞要保持氧化还原状态,也必须时刻都有fNrf2进入细胞核促使抗氧化基因表达,所以认为 Li等[32]对 Nrf2/Keap1信号通道机制的研究更为合理。
2 DNA损伤及其反应机制
动物体内DNA经常受到来自体内外各种因素的刺激,例如:外源性的高温高压、紫外线、射线、重金属、强氧化剂、强酸和强碱等物理化学因素和内源性ROS、酸碱不平衡及DNA在复制和传递过程中出现的错误等,这些因素都将导致DNA损伤。DNA损伤的形式有多种,如DNA双链结构破坏(DSBs)[37]、DNA 链断裂、碱基或碱基对被切除或替换等。此外,DNA损伤会引发多种细胞反应,包括DNA修复、细胞周期延迟或阻滞、细胞凋亡[38]等。由于DNA损伤的形式有多种,因而其损伤反应的机理也各不相同,由于DSBs出现在多种原因导致的DNA损伤中[39],本文主要介绍了DSBs诱发的反应。
DSBs是DNA损伤中最具有致命性作用的损伤之一,若DSBs没有被成功修复,不仅危及细胞的自我更新和分化能力,而且导致染色体组不稳定和疾病[39]。在哺乳动物细胞内,当MRN复合体(由 Mre11、Rad50和 Nbs1组成)感应到 DSBs时,立即激活运动失调性毛细血管扩张症突变蛋白(ataxia telangiectasia mutated,ATM),磷脂酰肌醇-3-激酶 β(phosphoinositide 3-kinaseβ,PI3Kβ)也是感应染色体结构完整性的一个重要感受器[40],PI3K相关激酶(PIKK)家族的成员参与组织DNA损伤反应[41-42]。其中ATM 能使细胞周期控制、DNA修复和染色体结构调节中的700多种蛋白质磷酸化,包括p53、细胞周期检测点激酶 2(Chk2)和组蛋白家族成员 X(H2AX)[43],并且有研究表明,ATM在氧化应激反应中也起到关键的作用[44]。被激活的ATM导致H2AX快速磷酸化形成γ-H2AX,从而在损伤处标定DNA损伤信号和积累修复蛋白,γ-H2AX能被小脑症蛋白(MCPH1)和DNA损伤调节蛋白1(MDC1)识别,MDC1通过磷酸酶和组蛋白交换因子的活动扩大依赖ATM的信号,与此同时也促进了其他DSBs信号和修复蛋白的积累,最终DNA损伤信号导致检查点蛋白激酶细胞周期检测点激酶1(Chk1)和Chk2被激活,Chk1和Chk2能修改细胞周期的部分机制使得细胞周期停止[45]。细胞在长期进化过程中产生细胞周期检测点(探测器包括Rad9和Rad17等)以保证细胞周期中DNA复制和染色体质量的机制,由于DNA损伤可以发生在细胞周期的任何时相,因而DNA损伤检查点又可分为:G1期、M 期和 S2期检查点[46]。若 G1期发生 DNA损伤,激活后的Chk1和Chk2可以磷酸化细胞分裂周期蛋白25A(Cdc25A),使其降解,即没有足够的Cdc25A清除细胞周期素依赖性激酶2(CDK2)上抑制其活性位点的磷酸基团,使得Cdc25A不能被激活,从而不能推动G1期向S期转换,引起细胞周期停滞[40];若S期发生DNA损伤,Chk1和Chk2抑制了CDK2活性,Cdc45不能组装到染色质上,Cdc45可以召集DNA聚合酶α,从而大大降低了DNA合成速度以延缓细胞周期[40,47];若 G2 期发生 DNA 损伤,Cdc25A 被泛素降解而不能激活细胞周期素依赖性激酶1(CDK1),且G2期检查点还会激活一条依赖p53的途径以停滞细胞周期[40,48]。细胞周期停滞或延缓后,通过碱基切除修复、核酸切除修复、重组修复和错配修复等方式修复受损DNA,如损伤不能修复或不能正确完成,则启动凋亡或非凋亡性死亡程序,清除有损伤或病变倾向的细胞[49]。值得注意的是,有研究报道,表皮生长因子受体(EGFR)与细胞外调节蛋白激酶(ERK)和蛋白激酶B(PKB)串联的信号对依赖ATM修复DNA损伤的反应有积极影响[50-51],并且修复DSBs有2条主要的途径,分别是同源重组(HR)和末端加入的非同源(non-homologous end-joining,NHEJ),这 2 条途径见的平衡是保持基因组稳定所必需的[52]。
3 ROS与DNA损伤之间的联系
大量研究表明,ROS能够引起氧化损伤,并攻击蛋白质和DNA,会引起包括DNA链断裂、DNA位点突变、DNA双链畸变和原癌基因与肿瘤抑制基因突变等形式的DNA损伤[6],例如,嗜中性粒细胞和巨噬细胞引起的ROS能直接导致DNA脱氨基和碱基氧化[53]。不仅如此,DNA损伤可以提升细胞内ROS水平[8],且 DNA损伤诱导的 ROS在调节细胞的死亡和自救中起到很重要的作用[9]。由此说明DNA损伤和ROS引起的氧化应激之间存在着一定的联系。
3.1 ROS引起的氧化应激导致DNA损伤
过多的ROS引起的氧化损伤会导致包括DNA链断裂、DNA位点突变、DNA双链畸变等形式的DNA损伤,但是与核DNA(nDNA)相比,线粒体DNA(mtDNA)更容易遭受氧化伤害[54]。有报道称,ROS对 mtDNA突变影响更大[55]。大多数mtDNA突变的形式是:胸腺嘧啶(T)转变为胞嘧啶(C),鸟嘌呤(G)转变为腺嘌呤(A)[56]。而ROS可使G氧化为8-氧鸟嘌呤(8-oxoG),而8-oxoG不再与C配对,反而与A配对,使得G转变为A[57]。此外,Fenton反应分解H2O2生成的高活性·OH能高效地损伤DNA,包括:DNA链断裂、DNA位点突变、DNA双链畸变。并且线粒体呼吸链产生的ROS导致的mtDNA损伤和突变能反过来使呼吸链功能失效,从而进一步促进ROS的产生[58]。由此说明,ROS引起的氧化应激与 DNA损伤之间有相互促进作用。
3.2 DNA损伤引起的ROS导致氧化应激
在细胞培养中,DNA损伤已被证明可以刺激细胞中 ROS 产生[59],如·、H2O2和·OH[60]。DNA损伤后的2 h内ROS量迅速增加,2.0~3.5 h逐渐减少,3.5~5.0 h内出现第2次迅速增加[61],但其机制仍不清楚。有研究表明,DNA损伤通过H2AX-还原型辅酶Ⅱ氧化酶1(Nox1)/Rac1通道诱导ROS产生,H2AX的超量表达和基因敲除都证明H2AX能调节ROS的产生[61],其原因可能是由于DNA损伤后导致H2AX被磷酸化的量增加,H2AX通过掩蔽14-3-3zeta[14-3-3zeta蛋白是14-3-3家族的一个成员,通常以二聚体的形式存在,在真核细胞中普遍存在,它能同时接受2个配体(如受体蛋白和激酶等),是信号传导通路中的重要接头蛋白[60]],激活 Rac1、GTP 酶和Nox1,从而引起细胞内 ROS的量增加。其中,Nox1是gp91(phox)(一个NADPH氧化酶的亚单位)的同源分子,其基因可在非噬细胞(上皮细胞和内皮细胞等)内表达,能被Rac1(细胞内重要的信号转导分子)激活。研究表明,减少Rac1的生成能减少依赖Nox1产生的ROS量[62]。
4 小结
综上所述,若ROS引起的氧化应激和各种原因诱导的DNA损伤超过了动物机体细胞自我修复的能力,将会对动物机体造成各种损伤,如肿瘤、癌症、细胞衰老甚至凋亡等,并且DNA损伤和氧化应激之间存在着必然的联系。但是一些细胞自我修复和抵抗各种应激与ROS和DNA损伤的机制尚不清楚,并且氧化应激和DNA损伤对机体细胞造成损伤的广泛性及其在动物生产中产生的影响需要做进一步的研究。
[1]REUTER S,GUPTA S C,CHATURVEDI M M,et al.Oxidative stress,inflammation,and cancer:how are they linked?[J].Free Radical Biology and Medicine,2010,49(11):1603-1616.
[2]PI J B,ZHANG Q,FU JQ,et al.ROS signaling,oxidative stress and Nrf2 in pancreatic beta-cell function[J].Toxicology and Applied Pharmacology,2010,244(1):77-83.
[3]KANNINEN K,WHITE A R,KOISTINAHO J,et al.Targeting glycogen synthase kinase-3βfor therapeutic benefit against oxidative stress in Alzheimer’s disease:involvement of the Nrf2-ARE Pathway[J].International Journal of Alzheimer’s Disease,2011,2011:985085.
[4]SYKIOTIS G P,HABEOS I G,SAMUELSON A V,et al.The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation[J].Current Opinion in Clinical Nutrition and Metabolic Care,2011,14(1):41-48.
[5]PANTANO C,REYNAERT N L,VAN DER VLIET A,et al.Redox-sensitive kinases of the nuclear factorκB signaling pathway[J].Antioxidants & Redox Signaling,2006,8(9/10):1791-1806.
[6]MEIRA L B,BUGNI J M,GREEN S L,et al.DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice[J].The Journal of Clinical Investigation,2008,118(7):2516-2525.
[7]DAS P M,SINGAL R.DNA methylation and cancer[J].Journal of Clinical Oncology,2004,22(22):4632-4642.
[8]ROWE L A,DEGTYAREVA N,DOETSCH P W.DNA damage-induced reactive oxygen species(ROS)stress response in Saccharomyces cerevisiae[J].Free Radical Biology and Medicine,2008,45:1167-1177.
[9]HAMANAKA R B,CHANDEL N S.Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes[J].Trends Biochemical Sciences,2010,35:505-513.
[10]BRAGADO P,ARMESILLA A,SILVA A,et al.Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation[J].Apoptosis,2007,12:1733-1742.
[11]LIU B,CHEN Y M,ST CLAIR D K.ROSand p53:a versatile partnership[J].Free Radical Biology and Medicine,2008,44:1529-1535.
[12]KLAUNIG JE,KAMENDULISL M.The role of oxidative stress in carcinogenesis[J].Annual Review of Pharmacology and Toxicology,2004,44:239-267.
[13]JOMOVA K,BAROS S,VALKO M.Redox active metal-induced oxidative stress in biological systems[J].Transition Metal Chemistry,2012,37(2):127-134.
[14]ST-PIERRE J,BUCKINGHAM J A,ROEBUCK S J,et al.Topology of superoxide production from different sites in the mitochondrial electron transport chain[J].The Journal of Biological Chemistry,2002,277(47):44784-44790.
[15]VALKO M,RHODES C J,MONCOL J,et al.Free radicals,metals and antioxidants in oxidative stress-in-duced cancer[J].Chemico-Biological Interactions,2006,160(1):1-40.
[16]KLAUNIG J E,WANG Z,PU X,et al.Oxidative stress and oxidative damage in chemical carcinogenesis[J].Toxicology and Applied Pharmacology,2011,254(2):86-99.
[17]VALKO M,IZAKOVIC M,MAZUR M,et al.Role of oxygen radicals in DNA damage and cancer incidence[J].Molecular and Cellular Biochemistry,2004,266:37-56.
[18]RICQUIER D,BOUILLAUD F.The uncoupling protein homologues:UCP1,UCP2,UCP3,StUCPand At-UCP[J].The Biochemical Journal,2000,345:161-179.
[19]NEDERGAARD J,RICQUIER D,KOZAK L P.Uncoupling proteins:current status and therapeutic prospects[J].EMBO Reports,2005,6(10):917-921.
[20]SUSKI J M,SCHÜNFELD P,BONORA M,et al.Uanosine diphosphate exerts a lower effect on superoxide release from mitochondrial matrix in the brains of uncoupling protein-2 knockout mice:new evidence for a putative novel function of uncoupling proteins as superoxide anion transporters[J].Biochemical and Biophysical Research Communications,2012,428(2):234-238.
[21]NABBEN M,HOEKSJ,BRIEDÉ JJ,et al.The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice[J].FEBS Letters,2008,582(30):4147-4152.
[22]BRAND M D,ESTEVEST C.Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3[J].Cell Metabolism,2005,2(2):85-93.
[23]AFFOURTIT C,CRICHTON PG,PARKER N,et al.Novel uncoupling proteins[J].Novartis Foundation Symposium,2007,287:70-80.
[24]AZZU V,BRAND M D.The on-off switches of the mitochondrial uncoupling proteins[J].Trends in Biochemical Sciences,2010,35(5):298-307.
[25]MCCUBREY J A,LAHAIR M M,FRANKLIN R A.Reactive oxygen species-induced activation of the MAP kinase signaling pathways[J].Antioxidants &Redox Signaling,2006,8(9/10):1775-1789.
[26]BYUN Y J,KIM SK,KIM Y M,et al.Hydrogen peroxide induces autophagic cell death in C6 glioma cells via BNIP3-mediated suppression of the mTOR pathway[J].Neuroscience Letters,2009,461(2):131-135.
[27]KANTHASAMY A G,KITAZAWA M,KANTHASAMY A,et al.Role of proteolytic activation of protein kinase Cδin oxidative stress-induced apoptosis[J].Antioxidants & Redox Signaling,2003,5(5):609-620.
[28]STEPKOWSKI T M,KRUSZEWSKI M K.Molecular cross-talk between the NRF2/KEAP1 signaling pathway,autophagy,and apoptosis[J].Free Radical Biology and Medicine,2011,50(9):1186-1195.
[29]WAKABAYASHI N,DINKOVA-KOSTOVA A T,HOLTZCLAW W D,et al.Protection against electrophile and oxidant stress by induction of the phase 2 response:fate of cysteines of the Keap1 sensor modified by inducers[J].Proceedings of the National Academy of Sciences of the United States of America,2004,101(7):2040-2045.
[30]OKAWA H,MOTOHASHI H,KOBAYASHI A,et al.Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity[J].Biochemical and Biophysical Research Communications,2006,339(1):79-88.
[31]LEVONEN A L,LANDAR A,RAMACHANDRAN A,et al.Cellular mechanisms of redox cell signalling:role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products[J].Biochemical Journal,2004,378:373-382.
[32]LI W G,KONG A N.Molecular mechanisms of Nrf2-mediated antioxidant response[J].Molecular Carcinogenesis,2009,48(2):91-104.
[33]PURDOM-DICKINSON S E,SHEVELEVA E V,SUN H P,et al.Translational control of nrf2 protein in activation of antioxidant response by oxidants[J].Molecular Pharmacology,2007,72(4):1074-1081.
[34]YAMAMOTO T,KYO M,KAMIYA T,et al.Predictive base substitution rules that determine the binding and transcriptional specificity of Maf recognition elements[J].Genes to Cells,2006,11(6):575-591.
[35]ZHANG D D,LO S C,SUN Z,et al.Ubiquitination of Keap1,a BTB-Kelch substrate adaptor protein for Cul3,targets Keap1 for degradation by a proteasomeindependent pathway[J].The Journal of Biological Chemistry,2005,280(34):30091-30099.
[36]YUAN X L,XU C J,ZUI P,et al.Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells[J].Molecular Carcinogene-sis,2006,45(11):841-850.
[37]NAGARIA P,ROBERT C,RASSOOL F V.DNA double-strand break response in stem cells:mechanisms to maintain genomic integrity[J].Biochimica et Biophysica Acta(BBA):General Subjects,2013,1830(2):2345-2353.
[38]蒋满荣.DNA损伤对哺乳动物细胞周期和凋亡的影响[D].博士学位论文.上海:中国科学院研究生院,2006.
[39]KHALIL A,MORGAN R N,ADAMS B R,et al.ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks[J].Cell Cycle,2011,10(3):481-491.
[40]KUMAR A,FERNANDEZ-CAPETILLO O,CARRERA A C.Nuclear phosphoinositide 3-kinaseβcontrols double-strand break DNA repair[J].Proceedings of the National Academy of Sciences of the United States of America,2010,107(16):7491-7496.
[41]BARTEK J,LUKAS J.DNA damage checkpoints:from initiation to recovery or adaptation[J].Current Opinion in Cell Biology,2007,19:238-245.
[42]HARRISON JC,HABER JE.Surviving the breakup:the DNA damage checkpoint[J].Annual Review of Genetics,2006,40:209-235.
[43]MATSUOKA S,BALLIF B A,SMOGORZEWSKA A,et al.ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage[J].Science,2007,316:1160-1166.
[44]SHILOH Y.ATM(ataxia telangiectasia mutated):expanding roles in the DNA damage response and cellular homeostasis[J].Biochemical Society Transactions,2001,29:661-666.
[45]YEUNG M,DUROCHER D.Engineering a DNA damage response without DNA damage[J].Genome Biology,2008,9(7):227.
[46]AGAMI R,BERRARDS R.Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage[J].Cell,2000,102:55-66.
[47]BUSINO L,DONZELLI M,CHIESA M,et al.Degradation of Cdc25A byβ-TrCP during S phase and in response to DNA damage[J].Nature,2003,426:87-91.
[48]LUKAS J,LUKAS C,BARTEK J.Mammalian cell cycle checkpoints:signalling pathways and their organization in space and time[J].DNA Repair,2004,3(8/9):997-1007.
[49]NOWSHEEN S,YANG E S.The intersection between DNA damage response and cell death pathways[J].Experimental Oncology,2012,34(3):243-254.
[50]GOLDING S E,MORGAN R N,ADAMS B R,et al.Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvⅢenhances DNA double-strand break repair in human glioma cells[J].Cancer Biology &Therapy,2009,8:730-738.
[51]MUKHERJEE B,MCELLIN B,CAMACHO C V,et al.EGFRvⅢand DNA double-strand break repair:a molecular mechanism for radioresistance in glioblastoma[J].Cancer Research,2009,69:4252-4259.
[52]BRANDSMA I,VAN GENT D C.Pathway choice in DNA double strand break repair:observations of a balancing act[J].Genome Integrity,2012,3(1):9.
[53]COUSSENS L M,WERB Z.Inflammation and cancer[J].Nature,2002,420:860-867.
[54]LIANG F Q,GODLEY B F.Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells:a possible mechanism for RPE aging and age-related macular degeneration[J].Experimental Eye Research,2003,76(4):397-403.
[55]ISHIKAWA K,TAKENAGA K,AKIMOTO M,et al.ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis[J].Science,2008,320:661-664.
[56]LI H,HONG Z H.Mitochondrial DNA mutations in human tumor cells[J].Oncology Letters,2012,4(5):868-872.
[57]WANG D,KREUTZER D A,ESSIGMANN J M.Mutagenicity and repair of oxidative DNA damage:insights from studies using defined lesions[J].Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis,1998,400:99-115.
[58]SHOKOLENKO I,VENEDIKTOVA N,BOCHKAREVA A,et al.Oxidative stress induces degradation of mitochondrial DNA[J].Nucleic Acids Research,2009,37(8):2539-2548.
[59]EVERT B A,SALMON T B,SONG B W,et al.Spontaneous DNA damage in Saccharomyces cerevisiae elicits phenotypic properties similar to cancer cells[J].Journal of Biological Chemistry,2004,279:22585-22594.
[60]吴东明.14-3-3zeta蛋白多克隆抗体的制备与鉴定[D].硕士学位论文.合肥:安徽大学,2007.
[61]KANG M A,SO E Y,SIMONS A L,et al.DNA damage induces reactive oxygen species generationthrough the H2AX-Nox1/Rac1 pathway[J].Cell Death and Disease,2012,3:e249.
[62]CHENG G J,DIEBOLD B A,HUGHES Y,et al.Nox1-dependent reactive oxygen generation is regulated by Rac1[J].Journal of Biological Chemistry,2006,281(26):17718-17726.
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际呼吸杂志(2019年4期)2019-03-12
中华老年多器官疾病杂志(2016年7期)2016-04-28
癌症进展(2016年10期)2016-03-20
西南军医(2016年6期)2016-01-23
中国医药生物技术(2015年4期)2015-12-26
医学研究杂志(2015年5期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
