
可控形貌的双金属纳米晶催化剂的研究进展*
2013-09-18 08:44张治平张亚文
大学化学 2013年5期
张治平 张亚文
(北京大学化学与分子工程学院稀土材料化学及应用国家重点实验室 北京100871)
随着经济的快速发展和人们生活水平的提高,绿色环保、高效节能正成为一种衡量社会是否能可持续健康发展的指标。多相催化剂与均相催化剂相比,具有高循环利用率和回收时能量的低消耗,因而在绿色化学、环境修复、生物质的高效转化、可再生新能源开发等领域日益引起人们的广泛研究兴趣。金属纳米催化剂,尤其是那些对反应物和产物具有适中的吸附脱附能力的贵金属,在环境污染治理、燃料电池、精细有机合成等多相催化反应中扮演极其重要的角色。比如,贵金属铂族元素(如Pt、Pd、Rh)是治理汽车尾气污染的三效催化剂中的活性金属成分,它们用于将废气中的CO和碳氢化合物氧化成二氧化碳、氮氧化物还原成氮气等[1]。但是贵金属在地壳中的丰度非常低,因此储量极其有限。近20年来,如何在减少贵金属用量的同时,保持甚至提高贵金属催化剂的性能以及循环利用效率成为一个研究的焦点。其中,将地壳中丰度较高、价格便宜的过渡金属引入到贵金属中,从而组成双金属纳米催化剂是解决贵金属的部分替代和减量化使用问题的一种可行方案[2-6]。
由两种不同金属组成的纳米催化剂因拥有优异的催化性能和较为廉价的成本,正成为无机化学、纳米科学、催化化学等多学科领域的交叉研究的一个前沿基础课题。由于双金属具有二组分的共同能带,因此,通过改变合金的组成,能够连续地改变颗粒表面的电子结构和几何构型;通过调节金属的d带位置与Fermi能级之间的距离,能够改变纳米颗粒对反应物和生成物的吸附能力,进而影响催化剂的活性、选择性和稳定性等[4]。例如,Pt常用作甲醇燃料电池的阳极催化剂,然而甲醇氧化过程中生成的CO在Pt上的吸附能力很强,使得Pt上的活性位点减少,从而影响了甲醇的进一步催化氧化。2011年,孙守恒课题组指出Pt-Pd合金纳米多面体体系拥有催化甲醇氧化(MOR)的双功能活性位点[5]。当在Pt纳米颗粒中引入Pd时,Pd能促进水的解离形成Pd-OH,Pt则催化甲醇氧化生成Pt-CO。Pd-OH与Pt-CO相互作用从而氧化生成CO2,并释放出Pt的活性位点,进而使得MOR催化反应持续进行。Sung Jong Yoo课题组在2012年报道了铂基双金属纳米催化剂Pt3M在氧气还原反应(ORR)中的应用[6],并指出催化活性存在下列关系:Pt<Pt3Zr<Pt3Co<Pt3Ni<Pt3Y。在Pt3Y 体系中,Y的电负性比Pt的电负性小,因此在形成Pt-Y金属键时,将有电子密度从Y原子流向Pt原子,从而改变Pt的电子结构和d带的深度,进而影响Pt位点对含氧物种的吸附脱附能力。
已有研究证明了双金属纳米催化剂的活性、选择性和稳定性不仅与颗粒的组成、尺寸有关,还与其纳米结构类型有关。例如,Eichhorn等人[7]指出在NO被H2还原生成N2的反应中,PtCu纳米合金颗粒拥有比纯铂更好的选择性,但活性比纯铂弱;而Cu@Pt核壳结构呈现出和纯铂一样的催化活性以及和PtCu纳米合金一样好的选择性,Pt@Cu颗粒在相同的条件下则体现出很低的催化活性。显然,发展一些能调控具有特定尺寸、组成和形貌的纳米颗粒的液相合成方法是该领域的迫切任务,也成为双金属纳米催化领域的前沿课题。
由于许多催化反应对金属纳米催化剂的结构非常敏感,因此本文首先将简单介绍双金属纳米晶的纳米结构类型,然后着重讲述近年来所发展的具有特定形貌的双金属纳米晶的合成方法,最后探讨这些纳米晶在电催化反应等多相催化反应中的研究现状和应用。
1 双金属纳米晶的结构类型
不同的原料和不同的制备方法将得到不同结构的纳米晶体。根据两种不同金属原子的混合和排布类型,可以将双金属纳米晶大致分成3类:合金结构(包括金属间化合物合金和固溶体合金)、核壳结构、多级结构[8],如图1所示。这些纳米结构根据合成条件,可外露各种晶面包括低密勒指数面、高密勒指数台阶面或它们的混合晶面。
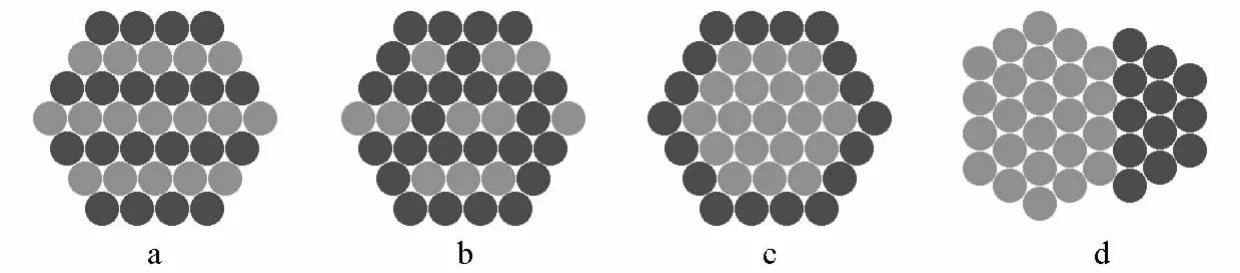
图1 双金属纳米晶的结构类型
1.1 合金结构
在液相合成反应中,当两种金属离子的前驱体同时被还原进而均匀成核生长时,将得到合金结构的纳米颗粒。具有合金结构的双金属纳米晶是具有特定原子几何排布的均匀混合物,金属原子之间有金属键形成。合金结构又分为金属间化合物(如图1a)和固溶体(如图1b)两种类型。金属间化合物合金中的各组分相互成键形成化合物,因此拥有不同于其组成元素结构的周期性、长程有序的结构;固溶体合金则各组分相互溶解,金属原子呈自由分布。在粉末XRD图中,纯金属的特征衍射峰将消失,出现不同于两种纯金属组分的合金衍射峰[9]。当两种金属的电负性不一样时,将有电子从电负性低的原子移向电负性高的原子,从而体现出不同于纯金属的电子结构,如d带位置的不同,进而影响表面金属原子在催化反应中对反应物种的吸附能力。一些合金体系会出现协同催化效应,即不同的金属原子负责吸附不同的物种。例如,Markovic等人发现PtRu纳米合金用于碱性氢燃料电池的阳极催化剂时,Pt将催化H2解离生成Had,Ru则提供位点吸附OH(OHad);然后邻近的Pt-Had与Ru-OHad相互作用生成水,从而表现出比纯铂更好的催化活性[10]。
1.2 核壳结构
当两种金属离子的前驱体具有不同的还原能力时,其中还原电势高的前驱体将优先被还原形成内核,之后电势低的金属离子被还原成金属原子并沉积在内核表面,最后形成核壳结构(如图1c)。当合成的纳米颗粒大于几十纳米时,XRD花样中将同时出现两种金属的特征峰;当纳米颗粒的尺寸小于一两个纳米时,由于衍射峰的严重宽化,可能造成金属特征峰的消失。核壳结构的纳米颗粒的催化性质主要取决于壳层金属的成分,呈现出与纯的壳层金属类似的催化效果。由于内外层金属间存在电负性差,因此壳层和内核之间的界面将存在电子的转移。当壳层金属只有1~2层时,这种电子的运动将影响壳层金属的催化活性和选择性。例如,Mavrikakis等人报导了由在过渡金属上覆盖1~2层Pt原子而组成的核壳纳米颗粒M@Pt,在CO选择性氧化反应中存在下列的催化活性次序:Ru@Pt>Rh@Pt>Ir@Pt>Pd@Pt>Pt>Au@Pt,其原因是内核基体的配体效应和不同原子间的晶格应力改变了壳层原子的电子结构[11]。比如,由于Au的电负性大于Pt的电负性,因此Pt原子的一部分d电子将转移到Au原子上,造成Pt原子的d带上移、与CO的结合力将加强,进而使得催化剂容易中毒。
1.3 多级结构
一种金属原子在由另一种金属所组成的基体表面上沉积时,具有3种生长模式,即层-层生长模式、薄膜-岛生长模式、岛生长模式。当金属N的N—N键能大于金属M与金属N间的M—N键能,或N的比表面自由能大于M的比表面自由能时,N会选择小岛生长模式,因为这样可以减小M与N之间的晶格应力和总的表面自由能。简单地说,当一种金属优先生成一定形貌的纳米晶体,另一种金属原子在晶体上采取“岛生长模式”时,将形成多级结构(如图1d)。比如,Tsung课题组最近合成了Pd-Rh的多级结构的纳米晶[12]。该课题组首先合成了Pd的纳米立方体,由于Rh—Rh的键解离能大于Rh—Pd的键解离能,因此,随后还原生成的Rh原子在Pd的晶体上将选择“岛状生长模式”,进一步形成多级结构的Pd-Rh纳米晶。多级结构纳米晶的特征和单纯地混合对应尺寸的金属纳米晶所形成的混合物的特征类似。多级结构纳米晶的XRD花样将出现和金属混合物一样的金属特征峰。一方面,多级结构的双金属纳米晶将拥有金属混合物类似的催化活性;另一方面,由于多级结构存在界面间相互作用,因此在某些体系中将拥有部分不同于金属混合物的催化特性,如在Pt表面生长出Ru小岛时,PtRu界面是甲醇氧化的优良活性位点[13]。当然,也有可能由于界面的存在,会导致部分活性位点的丧失和催化活性的减弱。
此外,若根据纳米晶体在三维方向上的伸展情况,又可以把双金属纳米晶的结构类型分为零维、一维以及二维结构,如表1所示。零维结构是指晶体在三维方向上的伸展情况在同一数量级,主要包括规则多面体、Wulff多面体[14]等;一维结构是指晶体在某一维度的伸展长度远大于另外两个维度的伸展长度,主要包括纳米线、纳米棒等;二维结构是指晶体在某两个维度的伸展长度远大于另外一个维度的伸展长度,主要有纳米板、纳米片和纳米带等。对于fcc结构的晶体,它的简单低指数面的表面活化能有下列关系:,因此纳米晶倾向于形成由{111}晶面组成的四面体或八面体。当有能稳定{100}晶面的选择性试剂存在时,纳米晶将形成立方体结构。相比于四面体和八面体,立方体的比表面积更小。因此,为了降低总的表面能,纳米晶更倾向于形成由多个{111}和{100}晶面组成的Wulff多面体。从结晶状态来讲,以上不同维度的双金属纳米结构有些是单晶状态,有些是多晶状态,而有些是多重孪晶状态(主要包括(111)孪晶界)[14-25]。
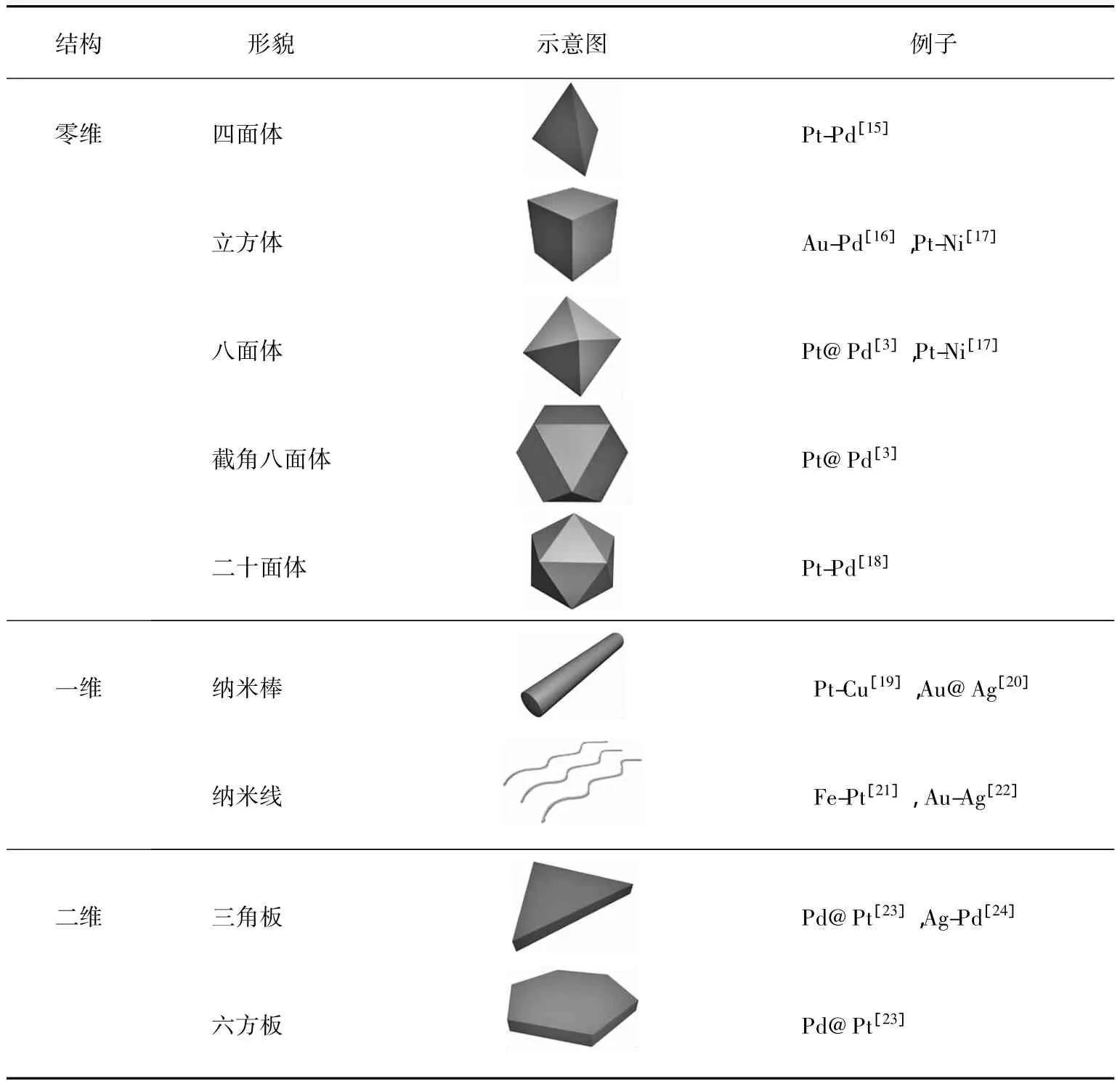
表1 纳米晶体常见的形貌
2 双金属纳米晶的可控合成方法
金属纳米催化剂的催化特性与其组成、尺寸、形貌等密切相关。因此,实现双金属纳米晶体的可控合成对深入研究其催化性质和探讨其催化机理具有重要的意义。一般来讲,纳米晶体的生长过程可以分为成核和生长两个阶段。然而,这两个阶段的热力学和动力学过程往往很难控制,而且成核和生长过程经常是连续的且难以分隔,因此纳米晶体的可控合成就变得很复杂。在整个合成过程中,反应温度的控制程序、环境压力的变化以及反应时间的长短都将影响晶体的形貌;另一方面,反应物的种类、浓度和还原电势,还原剂的选择,以及晶面选择试剂的加入也都将影响反应物的还原速率和纳米晶体的成核生长过程[25]。本节将概括性地介绍几种常用的液相控制合成方法。
2.1 一步合成法
两种金属离子的前驱体在一定的反应条件下可以同时被还原,进而成核生长成合金结构、核壳结构或多级结构。一步合成法就是指一次性混合所有的反应试剂,然后置于特定的反应条件下,最终得到双组分的金属纳米颗粒。这种合成方法具有操作少、流程简单的特点,因此是合成双金属纳米晶体的常用方法。根据反应途径和反应条件的不同,一步合成法可以分为溶剂热法和油相高温法等方法。
2.1.1 溶剂热法
将反应物浸入到水或有机溶剂中,而后在特定温度的密闭容器中反应一段时间,能有效调控纳米材料的结构和晶相。这种溶剂热反应具有独特的优势和广阔的应用前景[26]。溶剂热条件下,整个体系处于高温高压的状态,溶剂处于亚临界状态,溶解性加强,因而反应物的活性大大提高,能够制备常压下难以得到的亚稳态物相。溶剂热的反应参数易于调节,且能够有效地制备结晶度较高、尺寸分布小的纳米晶体。另外,纳米晶体的生长方式还与晶面选择试剂有关。因此,在溶剂热过程中,可以加入晶面选择试剂来控制纳米颗粒的形貌。不同晶面拥有不同的原子排布,有机试剂和无机离子在不同的晶面上拥有不同的吸附能力,因而可以控制纳米颗粒沿着特定的方向生长。比如,张亚文课题组利用水热合成法制备出了Pt-Pd的纳米四面体和纳米立方体[15]。草酸根离子能稳定Pt-Pd纳米颗粒的{111}晶面,有利于合成Pt-Pd纳米四面体。而碘离子和溴离子则能够选择性吸附在Pt-Pd纳米颗粒的{100}晶面,降低该晶面的表面自由能,因而适用于高效地制备Pt-Pd纳米立方体。利用PVP温和的还原能力和KBr的特定吸附作用,在160℃下可以还原K2PtCl4和CuCl2,并生成具有丰富台阶面原子的凹面Pt-Cu纳米颗粒[27]。李亚栋课题组在150℃利用苯甲醇的溶剂热作用、苯甲酸的晶面生长抑制剂作用与还原作用,合成了水溶性的Pt-Ni合金八面体[28]。Carpenter等人在不加包覆试剂的情况下,利用N,N-二甲基甲酰胺(DMF)的溶剂热法,合成了Pt3Ni合金的纳米立方体,其中DMF既作为溶剂又作为温和的还原剂[29]。
2.1.2 油相高温法
在高沸点溶剂充当反应介质时,金属前驱体在分解温度以上能够爆发性成核,因此成核和生长阶段能够较好地分开,便于制备尺寸均一、分散性好的纳米晶体。在温度升高之前,可以对反应体系进行抽真空;在温度升高和稳定的阶段,可以充入惰性气体隔绝氧气。因此,对于氧气敏感的制备反应过程,油相高温法拥有水热反应所没有的优势。高沸点溶剂多为长烷基链表面活性剂,包覆纳米颗粒的能力强,因此在应用于催化反应之前需要消除表面的配体,才能使得催化活性位点被释放。比如,Dimitrov等人在十四烷二醇、四辛基溴化铵、正十二硫醇以及油胺存在的条件下,用十八烯在220~230℃下还原Pt(acac)2(acac为乙酰丙酮)和 Cu(acac)2,合成出 Pt-Cu的纳米立方体[30]。基于二醇能将 Pt(acac)2还原成Pt颗粒,Fe(CO)5高温能分解产生Fe颗粒,以及油酸、油胺的混合物能稳定纳米颗粒,孙守恒等人制备出单分散的FePt纳米颗粒[2]。同样,Weller等人在金刚烷甲酸存在的条件下制备出高结晶度的Pt3Co纳米晶[31]。孙守恒等人在三辛基膦和油胺介质中,在260℃下将Co(acac)2、PdBr2还原成单分散的CoPd纳米颗粒[32]。作者指出油胺既是溶剂又是还原剂,而三辛基膦不仅仅能稳定晶面,更能控制合金的成核和生长过程。由于三辛基膦的d轨道是空的,它将参与表面金属原子的反馈π键,因此三辛基膦是比油胺更强的配体,在反应过程中,将优先生成三辛基膦的Co配合物和Pd配合物。
2.2 多步合成法
多步合成法操作虽较为复杂,但可以制备出许多一步法难以得到的纳米晶结构,因而具有巨大的发展空间。小尺寸的单金属纳米晶体可以充当制备双金属纳米晶体的“硬模板”。若将一种金属原子沉积到另一种半径相近的金属原子上,吸附层会在衬底表面形成(1×1)结构,即每个吸附原子占据一个衬底单胞,衬底将自身的晶体结构、取向和晶格常数施加到吸附层。此外,还可以利用一种金属离子置换出另一种金属离子形成独特的纳米结构。对合成后的双金属纳米颗粒进行后处理,可以得到各种组成和不同形貌的纳米颗粒。晶种法、Galvanic置换法和化学腐蚀法都属于常用的多步合成法。
2.2.1 晶种法
晶种法常被用于制备核壳结构或多级结构的双金属纳米晶。均相成核所需要的活化能大于生长所需的活化能。因此,当溶液中含有预先加入的纳米晶种时,第2种金属离子被还原后将在晶种表面异相成核生长。这种生长方式拥有保形外延生长和非保形外延生长两种生长模式。不同的生长模式主要取决于金属离子的还原速度大小、表面自由能的大小、金属键能的大小、晶面稳定试剂的选择等因素。比如,HAuCl4在Pd立方体晶种存在的条件下被抗坏血酸还原生成核壳结构的Pd@Au晶体;而在更温和的还原剂柠檬酸的条件下则形成Pd-Au的多级结构[33]。特定形貌的晶种为第2种金属的外延生长提供了特定的晶面并影响最终产物的形貌。比如,Yang等人利用Pt的立方体晶种控制了Pd、Au的外延生长方式[3]。Au的晶格常数与Pt的晶格常数相差比较大(4.08%的晶格失配度),因而Au在Pt的表面呈各向异性生长,从而形成多级结构;而Pd和Pt的晶格匹配度好(0.77%的晶格失配度),因此Pd在Pt晶种表面外延生长形成Pt@Pd核壳结构。加入NO2可以调节Pd沿着Pt立方体<100>和<111>方向的生长速度,最终形成Pt@Pd八面体或截角八面体纳米颗粒。
2.2.2 Galvanic置换法
在实际生活中,常用电镀方法来保护特定的金属。在双金属纳米晶体的合成中,Galvanic置换法也是通过牺牲一种金属,而将另一种金属原子沉积到原有的金属纳米颗粒表面,从而合成形貌较为复杂的双金属纳米结构。比如,Mikhlin等人[34]先将还原性比较强的金属Pd制成具有特定构型的纳米颗粒,然后将其放在氧化电势更高的金属离子Au3+溶液中,Au3+将被还原成零价金属,从而沉积在Pd的纳米颗粒表面,进而形成具有核壳结构的Pd-Au纳米颗粒。还原性强的纳米金属在某种程度上起着模板的作用。利用Galvanic置换法还可以合成凹面结构或中空结构的双金属纳米结构。比如,2009年,郑南峰课题组利用Pt(acac)2/Pd(acac)2共还原合成了具有中空结构的Pd-Pt纳米立方颗粒[35];Sotiropoulos等人将沉积了Cu、Ni、Co的玻碳电极浸入到K2PtCl4的盐酸溶液中制备出了Pt(Cu)、Pt(Ni)、Pt(Co)的电极[36]。虽然需要损失一定的金属,但Galvanic置换法不失为一种合成具有复杂结构的双金属纳米颗粒的好方法。
2.2.3 化学腐蚀法
化学腐蚀存在于日常生活中,如钢铁的生锈过程能够显著地改变物质的组成、形貌和稳定性,而这种腐蚀过程往往是混乱无序和难以控制的。但是若能有效地控制化学腐蚀的部位和反应进程,那么化学腐蚀就有望成为一种双金属纳米颗粒的可控合成方法。通过这种方法,可以制备出组成可调、形貌奇特的纳米颗粒。比如,2012年,李亚栋课题组报道了用可控的化学腐蚀法制备具有特定形态的Pt-Ni纳米催化剂[37]。Pt-Ni纳米合金表面上的Ni原子在加入丁二酮肟的水中能被氧气氧化成Ni2+,Ni2+与丁二酮肟形成配合物沉淀,进而促使反应从Ni向Ni2+进行。Pt2+由于不与丁二酮肟形成配合物,因此合金中的Pt难以被空气氧化。因此,在化学腐蚀中,Pt-Ni纳米合金的Ni逐渐减小,Pt基本不变,形成凹面的Pt-Ni纳米结构。若反应开始前的Pt-Ni合金中Ni的含量越多,则合金被腐蚀得越厉害,凹面趋势越大。同时,随着化学腐蚀进度的不同,所得到的纳米颗粒的凹面率也不同。化学腐蚀首先从顶点和边缘的Ni原子开始,随着Ni原子被氧化,不被空气氧化的Pt原子开始沉积和聚集,形成新的台阶和顶点。因此,不同凹面结构的形成归结于特定位点原子的优先腐蚀次序。凹面结构的Pt-Ni合金具有很大的比表面积和低配位数的原子,因此与一般的Pt-Ni合金相比,它具有更高的催化活性。此外,Yang等人利用化学腐蚀法合成了笼-铃状的Pt-Ru纳米晶体。作者首先合成了Pt@Ag@Ru的纳米结构,利用二(4-磺酸基苯基)-苯基磷酸盐对Ag(Ⅰ)/Ag(0)的强结合作用促进中间层的Ag被氧化,并扩散到壳层外形成笼-铃状的Pt-Ru纳米晶体[38]。因此,通过可控的化学腐蚀可以合成具有高比表面积的双金属纳米催化剂。
3 双金属纳米晶的催化性质
双金属纳米催化剂拥有可调的组成、可控的形貌和可变的电子结构,因此在溶液电催化反应中有着广泛的应用,如甲醇电氧化、氢气电氧化、氧气还原反应等。同时,它们还常被用于催化固液气相化学反应,如CO氧化、NOx还原、石油重整等。下面简单介绍双金属纳米晶在燃料电池阴阳极等多相催化反应中的应用探索现状。
3.1 电化学催化反应
3.1.1 甲醇氧化反应(MOR)
甲醇燃料电池具有很高的能量密度,而且其中的质子交换膜具有比较高的电导率。因此,甲醇燃料电池是一种有潜在应用价值的电池,也日益受到化学和材料研究者的青睐。过渡金属,尤其是Pt基金属,常用作燃料电池阴阳极反应的催化剂。目前在实验研究中,电催化的甲醇氧化反应(MOR)常作为评价金属催化剂综合性能的探针反应。它的电极反应为:

该阳极反应可以进一步分解为[39]:

Pt基的纳米颗粒常用于催化甲醇的氧化,Pt能够很好地催化反应(2)~(5),从而生成Pt-CO。但是Pt在较高的电势时才能生成Pt-OH[5],因此反应(6)是MOR反应的速率控制步骤。一方面,在低电势时,反应(6)进行得很缓慢,使得反应(7)进行缓慢;另一方面,CO在Pt上的吸附能力很强,以至于随着反应的进行,Pt上的吸附位点将减少,最终导致MOR进行得迟缓。Pt-Ru合金是甲醇燃料电池的高效催化剂[13]。Ru在较低的电位下能促进反应(6)的进行形成Ru-OH,然后随着反应(7)的进行,Pt重新获得吸附位点促进了甲醇的氧化进程。
Pt基合金中不同的晶面拥有不同的原子排布和电子结构,如含有不同数量的台阶原子和扭结原子。Pt基纳米合金的{100}晶面拥有比{111}晶面更低的吸附CO的起始电位,因此Pt基合金的{100}晶面对于加快甲醇的氧化具有更好的效果;但是CO对该晶面具有更强的毒害性,因此{100}晶面的催化持续性更差[15]。另一方面,催化剂的活性不仅与暴露的晶面有关,还取决于颗粒的形貌。2011年,张亚文课题组发现在甲醇电氧化过程中,Pt-Pd的二十面体纳米晶体比Pt-Pd的四面体纳米晶体具有更高的催化活性,Pt-Pd合金的二十面体比Pt-Pd的四面体、铂黑、Pt/C拥有更正的氧化电势和更高的氧化电流[18]。作者推测虽然这两种纳米颗粒的组分相近,而且都是暴露的{111}晶面,但是相比于四面体,Pt-Pd的二十面体的{111}晶面之间的距离更大,拥有更多的晶体缺陷,这些都导致了它们对不同物种吸附能力的改变。因此,不同形貌的Pt-Pd合金在甲醇电氧化过程中表现出不同的催化性能。
3.1.2 氧气还原反应(ORR)
聚合物电解质膜燃料电池有望成为化石燃料的代替能源之一,然而其大规模应用主要受阴极反应即氧气还原反应(ORR)的动力学限制,包括阴极过电位的存在、阴极反应过程中催化剂的流失等问题[40]。催化剂要有足够的活性活化O2,同时要有足够的惰性防止自身被氧化。相对于Fe、Co、Ni、Ag、Au等金属,Pt拥有活性较高的位点和对OH、O等阴极中间产物较为适中的吸附能和脱附能,因此它是ORR反应的优良的催化剂[41]。由于Pt是一种贵金属,而且Pt在催化过程中流失严重,因此寻找一种活性高且稳定性好的Pt基合金以代替纯Pt就成为燃料电池领域中的一个研究热点。如当Pt与3d过渡金属形成合金时,不仅能减少催化剂的成本,更能通过电子的转移来改变表层Pt原子的d带电子结构[42]。一般认为,对于氧气还原反应有如下两个机理[41]:
机理一:

机理二:

机理一与机理二的最大不同点在于吸附氧原子O*的产生方式。机理一认为氧气分子在金属表面直接解离成氧原子,而机理二认为生成氧原子之前需要生成过氧氢。当电位在0.8V以下,机理二占主导作用,因为电位低时阴极容易吸附氢离子从而生成过氧氢。ORR的反应活性很大一部分取决于生成过氧氢时的能量变化和生成水时的动力学过程。当催化剂吸附O2、O的能力强时,脱附OH的能力就弱。OH的脱附是ORR的决速步。利用上述两个机理可以预测出阴极反应速率和氧吸附能将成“火山形状”的关系。这对于表层原子为Pt的Pt基合金同样适用。在Pt基合金中,前过渡金属的电子有部分转移到Pt原子上,使得Pt的d带中心加深,与OH的相互作用变弱,更容易生成水。比如,Greeley等人发现Pt3Sc和Pt3Y拥有比纯Pt更高的活性(用Pt3Sc做阴极催化剂时半波电位向正方向平移了20mV,而Pt3Y则正移了60mV),同时拥有相对于其他合金Pt3M更好的稳定性[43]。对于ORR反应,Pt基合金的催化活性不仅与组成有关,还与暴露的晶面密切相关。不同晶面的原子拥有不同的配位情况,配位数的不同导致金属d带电子填充度不同,进而导致吸附能力不同。比如,Markovic等人发现Pt3Ni拥有很高的ORR催化活性,且不同晶面间存在下列活性大小关系:Pt3Ni(100)<Pt3Ni(110)≪Pt3Ni(111)[40]。
3.2 固液气相催化反应
3.2.1 CO选择性氧化(CO-PROX)
质子交换膜燃料电池能够高效地将氢气的化学能转化为电能,因此在移动设备中有广泛的应用前景[44]。然而氢气主要来源于石油中碳氢化合物的重整,因此氢气原料中常含有一定量的CO。CO对于阳极材料有明显的毒害作用,Pt材料可接受的CO含量小于0.001%。因此,如何高效净化氢气中的CO以防止CO毒化燃料电池或反应中使用的金属催化剂就成为一个挑战性课题。氢气原料一般先经过水汽重整降低CO的含量,但是受到水汽重整反应的热力学限制,CO的含量只能降至0.1% ~1%(体积分数)[45]。若需要进一步减低CO的含量,就需要进行CO甲烷化或CO选择性氧化。下面将简单介绍双金属纳米催化剂用于CO选择性氧化的一些研究进展。
CO选择性氧化可以“深度净化”燃料气体,即往水汽重整后的原料中通入适当的空气,在催化剂的作用下,选择性氧化CO而不氧化氢气。双金属在CO-PROX反应中的催化作用已引起不少课题组的研究兴趣。比如,Aksoylu等人在研究Pt3Sn体系时,发现在吸附位点下方的Sn加强了CO的吸附,而在表面上的Sn起到弱化CO吸附能力的作用[46]。Takayuki Komatsu等人则发现Pt3Co/SiO2、PtCu/SiO2在温度为453K时拥有比Pt/SiO2、Pt/Al2O3更高的活性[47]。夏幼南课题组发现相对于Pd的纳米立方体和商业化的Pt/C催化剂,Pd-Pt纳米笼表现出更高的活性和更好的选择性[44]。Pt基的其他合金纳米材料,如 Pt-Ni、Pt-Re 等,同样得到不少课题组的关注[48]。
Mavrikakis课题组在研究M@Pt(M=Ru、Rh、Ir、Pd、Au)体系时,提出了一种关于在富氢条件下COPROX的反应机理[11],该机理可以分成两部分:
吸附氧原子(O*)的生成:

吸附氧原子(O*)的消耗:

PROX的反应活性和选择性取决于两个因素:1)O*是吸附氧分子直接解离生成的,还是经过生成过氧氢后才解离生成的;2)在消耗O*的过程中生成CO2和生成H2O的相互竞争[11]。在富氢环境下,氧气更趋向于生成过氧氢,而后解离成氧原子。对于M@Pt,CO*和O*生成CO2时所需的活化能小于O*与H*生成OH*时所需要的活化能,因此,O*的消耗过程主要由生成CO2来完成,这使得M@Pt具有较好的选择性。在M@Pt体系中,由CO和O2生成CO2所需的活化能有如下关系:Ru@Pt<Rh@Pt<Ir@Pt<Pd@Pt<Pt<Au@Pt,因此,PROX 的活性关系正好与之相反。
3.2.2 碳碳偶联反应
双金属纳米材料不仅用于固气催化反应,而且还被广泛地应用于液相有机合成反应,如Fe-Ru体系能够有效地选择性催化还原环己烯酮[49]、Ni@Pd体系能有效地催化Sonogashira碳碳偶联反应[50]等。碳碳偶联反应是一类重要的碳增反应,包括Sonogashira-、Heck-、Ullmann-、Suzuki-偶联等,在医药、化学、生化等众多行业中占有重要的地位。它们常常利用Pd的配合物作为催化剂、选择性有机试剂作为溶剂。然而,均相催化往往难以回收催化剂并且常常带来环境污染问题,因此如何在水相中进行该类合成反应是一个研究热点。研究表明[51-53],在催化过程中真正起作用的组分是零价的Pd,这使得在水相中利用Pd基的纳米颗粒催化碳碳偶联反应成为可能。Pd的纳米颗粒可以有效地起到催化的作用,但是小尺寸造成的团聚问题往往成为限制其催化效果的关键因素之一。利用适当的载体可以有效地分散催化剂和稳定催化剂的构型。Pd/C能在水作为介质时有效地催化卤代酚进行Suzuki-Miyaura偶联反应[54]。Pd/ZrO2也被证明能够有效地催化卤代芳烃在水溶剂中进行碳碳偶联反应[55]。然而,这些偶联反应只在纳米颗粒表面上进行而使得剩余的大部分Pd被浪费,因此,如何提高Pd的利用率和降低反应中Pd的用量就成为一个挑战性问题。向Pd的体系中引入铁系元素,不仅能降低催化剂成本,而且还能通过电子结构的调节来改变催化剂的活性和选择性。如Hyeon课题组利用三辛基镍的分解温度低于三辛基钯的分解温度,制备出能催化Sonogashira偶联反应的Ni@Pd纳米颗粒[50]。如Pd-Co的中空纳米微球能起到催化Sonogashira偶联反应的作用[56]。又如Pd-Fe体系中的Fe能够促进Pd活性位点的分散,因此拥有比Pd更高的催化活性和更好的稳定性,同时利用Fe的磁性有利于催化剂的回收;相比于实心的Pd-Fe体系,中空的Pd-Fe体系表现出更高的催化活性,因为后者具有更低的密实度和更多数量的活性位点[57]。
4 总结与展望
将一种金属引入到一种贵金属体系,不仅能节省贵金属用量、降低催化剂成本,而且还能通过组成的改变、原子排布的控制、电子结构的调节来改善催化效果。通过一步法、多步法可以合成具有特定形貌的双金属纳米颗粒。双金属纳米晶是燃料电池阴阳极反应的高效催化剂,不仅能催化甲醇氧化、甲酸氧化、氢气氧化等众多的阳极反应,而且还能大幅改善阴极的氧气还原反应的超电势高、反应速率太慢的问题。同时,双金属纳米晶因具有良好的催化稳定性和循环回收性,也是石油重整、汽车尾气治理以及包括碳碳偶联在内的众多有机合成反应等多相催化反应的关键催化材料。
近年来,双金属纳米催化剂的可控合成和催化性能的研究已取得了不小的进展,但仍处于初步阶段,还需要在以下几方面加强研究:目前的工作大多是基于铂、钯、金的体系展开的,而其他双金属体系还存在许多空白;特定晶面特别是具有高指数台阶面的双金属纳米结构的控制合成的原理还远未研究透彻;催化反应过程中催化剂的形貌变化和组分偏析的研究仍有待突破;活性与选择性、稳定性间的矛盾仍有待平衡解决;纳米颗粒上的表面活性剂对催化性质的影响及其有效去除方法都需要进行深层次探索。我们相信,随着对双金属纳米晶控制合成化学和催化化学的深入研究,我们将能更好地调控纳米催化剂的组成、形貌、尺寸,并为实现低成本、大规模地合成高性能的双金属纳米催化剂奠定坚实的基础,进而为实现社会经济的绿色环保、节能减排的目标贡献一份力量。
[1]Gandhi H S,Graham G W,McCabe R W.J Catal,2003,216:433
[2]Sun SH,Murray C B,Weller D,et al.Science,2000,287:1989
[3]Habas SE,Lee H,Radmilovic V,et al.Nat Mater,2007,6:692
[4]Xin H,Holewinski A,Schweitzer N,et al.Top Catal,2012,55:376
[5]Liu Y,Chi M F,Mazumder V,et al.Chem Mater,2011,23:4199
[6]Hwang SJ,Kim SK,Lee JG,et al.J Am Chem Soc,2012,134:19508
[7]Zhou SH,Varughese B,Eichhorn B,et al.Angew Chem Int Ed,2005,44:4539
[8]Wang D S,Li Y D.Adv Mater,2011,23:1044
[9]Toshima N,Yonezawa T.New J Chem,1998,22:1179
[10]Strmcnik D,Wang C,Markovic N M.Nat Chem,2013,7
[11]Nilekar A U,Alayoglu S,Eichhorn B,et al.J Am Chem Soc,2010,132:7418
[12]Sneed B T,Kuo C H,Brodsky C N,et al.J Am Chem Soc,2012,134:18417
[13]Hepel M,Dela I,Hepel T,et al.Electrochim Acta,2007,52:5529
[14]Marks L D.Rep Prog Phys,1994,57:603
[15]Yin A X,Min X Q,Zhang Y W,et al.J Am Chem Soc,2011,133:3816
[16]Fan F R,Liu D Y,Wu Y F,et al.J Am Chem Soc,2008,130:6949
[17]Zhang J,Yang H Z,Fang JY,et al.Nano Lett,2010,10:638
[18]Yin A X,Min X Q,Zhu W,et al.Chem Commun,2012,48:543
[19]Liu Q S,Yan Z,Henderson N L,et al.J Am Chem Soc,2009,131:5720
[20]Xiang Y J,Wu X C,Liu D F,et al.Langmuir,2008,24:3465
[21]Wang C,Hou Y L,Kim J M,et al.Angew Chem Int Ed,2007,46:6333
[22]Hong X,Wang D S,Yu R,et al.Chem Commun,2011,47:5160
[23]Lim B,Wang JG,Camargo P H C,et al.Nano Lett,2008,8:2535
[24]Lee C L,Tseng C M,Wu R B,et al.Colloids Surf Physicochem Eng Asp,2009,352:84
[25]Gu J,Zhang Y W,Tao F.Chem Soc Rev,2012,41:8050
[26]Walton R I.Chem Soc Rev,2002,31:230
[27]Yin A X,Min X Q,Zhu W,et al.Chem Eur J,2012,18:777
[28]Wu Y E,Cai SF,Wang D S,et al.J Am Chem Soc,2012,134:8975
[29]Carpenter M K,Moylan T E,Kukreja R S,et al.J Am Chem Soc,2012,134:8535
[30]Xu D,Bliznakov S,Liu Z P,et al.Angew Chem Int Ed,2010,49:1282
[31]Shevchenko E V,Talapin D V,Rogach A L,et al.J Am Chem Soc,2002,124:11480
[32]Mazumder V,Chi M F,Mankin M N,et al.Nano Lett,2012,12:1102
[33]Lim B,Kobayashi H,Yu T,et al.J Am Chem Soc,2010,132:2506
[34]Belousov O V,Belousova N V,Sirotina A V,et al.Langmuir,2011,27:11697
[35]Huang X Q,Zhang H H,Guo C Y,et al.Angew Chem Int Ed,2009,48:4808
[36]Papadimitriou S,Armyanov S,Valova E,et al.J Phys Chem C,2010,114:5217
[37]Wu Y E,Wang D,Niu Z,et al.Angew Chem Int Ed,2012,51:12524
[38]Liu H,Qu J L,Chen Y F,et al.J Am Chem Soc,2012,134:11602
[39]Markovic N M,Ross P N.Surf Sci Rep,2002,45:117
[40]Stamenkovic V R,Fowler B,Mun B S,et al.Science,2007,315:493
[41]Norskov J K,Rossmeisl J,Logadottir A,et al.J Phys Chem B,2004,108:17886
[42]Stamenkovic V R,Mun B S,Arenz M,et al.Nat Mater,2007,6:241
[43]Greeley J,Stephens I E L,Bondarenko A S,et al.Nat Chem,2009,1:552
[44]Zhang H,Jin M S,Liu H Y,et al.Acs Nano,2011,5:8212
[45]Park E D,Lee D,Lee H C.Catal Today,2009,139:280
[46]Gulmen M A,Sumer A,Aksoylu A E.Sur Sci,2006,600:4909
[47]Komatsu T,Tamura A.J Catal,2008,258:306
[48]Yu W T,Porosoff M D,Chen JG G.Chem Rev,2012,112:5780
[49]Andanson JM,Marx S,Baiker A.Catal Sci& Technol,2012,2:1403
[50]Son SU,Jang Y,Park J,et al.J Am Chem Soc,2004,126:5026
[51]Amatore C,Jutand A,Mbarki M A.Organometallics,1992,11:3009
[52]Ozawa F,Kubo A,Hayashi T.Chem Lett,1992,2177
[53]Beletskaya I P,Cheprakov A V.Chem Rev,2000,100:3009
[54]Sakurai H,Tsukuda T,Hirao T.J Org Chem,2002,67:2721
[55]Monopoli A,Nacci A,Calo V,et al.Molecules,2010,15:4511
[56]Li Y G,Zhou P,Dai Z H,et al.New J Chem,2006,30:832
[57]Li H,Zhu Z H,Li H X,et al.J Colloid Interface Sci,2010,349:613
猜你喜欢
火炸药学报(2022年5期)2022-11-04
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
物理学报(2018年22期)2018-12-18
石油化工建设(2018年2期)2018-07-11
