
巴特日七味丸中大叶茜草素与硫化汞的测定
2013-09-17 01:32许晓洁王玉华
中成药 2013年9期
许晓洁, 王 伟, 王玉华*
(1.内蒙古医科大学药学院,内蒙古呼和浩特010110;2.内蒙古自治区食品药品检验所,内蒙古呼和浩特010020)
蒙成药“巴特日七味丸”收载在《中华人民共和国卫生部药品标准》[1](蒙药分册)第79页,处方由草乌叶、诃子、茜草、多叶棘豆、黑芸香、人工麝香、银朱等七味药材组成。在对巴特日七味丸质量控制方法的研究过程中,参照《中国药典》2010年版[2]制草乌项下方法对草乌叶中双酯型生物碱的测定方法进行了研究,因生物碱的量太低,未能建立定量测定方法;参考文献报道[3],采用HPLC法对诃子进行定量测定方法的研究,因共存成分的干扰大,未能建立测定方法;黑芸香是蒙医用药材,缺少化学成分的研究工作基础。本实验用HPLC法测定了茜草中大叶茜草素的量;用容量法测定了银朱中HgS的量。本研究对巴特日七味丸的质量控制具有意义。
1 仪器与试药
1.1 仪器 双光束紫外可见分光光度计TU—1901(上海精科实验有限公司);高效液相色谱仪P230(大连伊利特分析仪器有限公司);电子天平AL204(梅特勒-托利多仪器有限公司);超声清洗仪AS3120(天津奥特赛恩斯仪器有限公司)。
1.2 试药 巴特日七味丸 (内蒙古蒙药股份有限公司,批号100619;内蒙古乌兰浩特中蒙制药有限公司,批号090401、091114、100416;内蒙古库伦蒙药厂,批号100804;内蒙古大唐药业有限公司,批号10454007、10454009)。
大叶茜草素对照品 (由中国药品生物制品检定所购入,批号为110884-200604);银朱 (由乌兰浩特中蒙制药有限公司提供);甲醇为色谱纯,水为高纯水,所用其他试剂均为分析纯。
2 方法与结果
2.1 高效液相色谱法测定大叶茜草素[4-5]茜草是巴特日七味丸的主要药味之一。茜草的主要化学成分为大叶茜草素、羟基茜草素等。本实验采用反相高效液相色谱法对大叶茜草素的测定方法进行了研究。经实验研究确定了提取条件和液相分离条件,并对HPLC法进行了方法学考察。该方法专属性和准确性好,可以作为巴特日七味丸质量控制的方法。
2.1.1 色谱条件 C18柱 (250 mm×4.6 mm,5 μm);流动相为甲醇-水 (85∶15);体积流量为1.0 mL/min;检测波长为250 nm,进样体积为20 μL。理论塔板数以大叶茜草素峰计算不低于4 000。
2.1.2 溶液的制备
2.1.2.1 供试品溶液的制备 依据《中国药典》2010年版“茜草”项下的含量测定方法,在此基础上进行单因素筛选确定了供试品溶液的制备方法。
取本品粉末约4.0 g,精密称定,置具塞锥形瓶中,精密加入甲醇100 mL,密塞,称定质量,暗处放置5~8 h,超声处理 (功率250 W,频率40 kHz)30 min,放冷再称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液50 mL,蒸干,残渣加甲醇-25%盐酸 (4∶1)混合溶液20 mL溶解,置水浴中加热水解30 min,立即冷却,加入三乙胺3 mL,混匀,转移至25 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.1.2.2 对照品溶液的制备 精密称取大叶茜草素对照品适量,加甲醇制成每1 mL含50μg的溶液,即得。
2.1.2.3 阴性对照溶液的制备 按照处方比例及制备工艺,取缺茜草的6味药材粉末制备阴性对照品。精密称取阴性对照品适量,按2.1.2.1项“供试品溶液的制备”方法制成阴性对照溶液。
2.1.3 线性关系考察 精密称取大叶茜草素对照品,用甲醇溶解并制成每1 mL含有大叶茜草素116.5μg的溶液,作为对照品溶液。精密吸取对照品溶液适量,加甲醇制成含大叶茜草素质量浓度为 11.65、23.30、34.95、46.60、58.25、69.90、116.5μg/mL等7组系列对照品溶液,分别精密吸取系列对照品溶液20μL,按“色谱条件”项下方法测定,记录各图谱的大叶茜草素峰面积积分值。以进样量对峰面积积分值进行回归分析。结果显示,大叶茜草素在0.233~2.33μg范围内与峰面积积分值呈良好的线性,回归方程为 Y=6 300.6X -267.53(r=0.997 8)。
2.1.4 精密度试验 取本品,按2.1.2.1项方法制备溶液,按2.1.1项下方法测定,记录色谱峰的积分值。6次测定结果的RSD为0.45%。
取对照品溶液,同法测定并记录色谱峰的积分值。6次测定结果的RSD为0.95%。
2.1.5 稳定性试验 取本品,按2.1.2.1项方法制备溶液,按2.1.1项下方法操作,于溶液制备后的0、0.5、1、2、4、6、8、24 h进样测定,记录色谱图。结果显示,供试品溶液在24 h内稳定。条件:在棕色瓶中避光放置。
2.1.6 重复性试验 取同一批号供试品 (内蒙古乌兰浩特中蒙制药有限公司,批号:090401)6份,按2.1.2.1项方法制备溶液,按2.1.1项下方法操作,记录色谱图,计算其平均质量分数为0.806 mg/g,RSD为2.00%。
2.1.7 专属性考察 按2.1.2.3项下方法制备溶液,按2.1.1项下方法操作,记录供试品溶液、阴性对照溶液、大叶茜草素对照品溶液的色谱图。结果显示,在对照品色谱峰保留时间对应的位置上,供试品溶液有色谱峰,阴性对照溶液没有色谱峰,方法的专属性好,见图1。
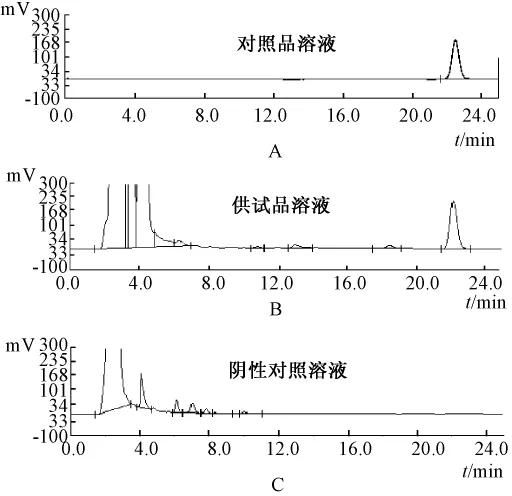
图1 大叶茜草素专属性试验色谱图Fig.1 Chromatograms of specificity tests of rubimaillin
2.1.8 加样回收试验 取已知含有量的巴特日七味丸样品 (内蒙古乌兰浩特中蒙制药有限公司,批号为090401,含有量为0.806 mg/g),称取9份,每份分别加入一定量大叶茜草素对照品,使之成为80%、100%和120%的加样回收试验样品,按2.1.2.1项方法制备溶液,按2.1.1项下方法操作,记录色谱图。加样回收试验结果见表1。表1的结果显示,80%、100%、120%回收试验结果的均值为99.0%,RSD为1.61%,表明方法的准确度好。
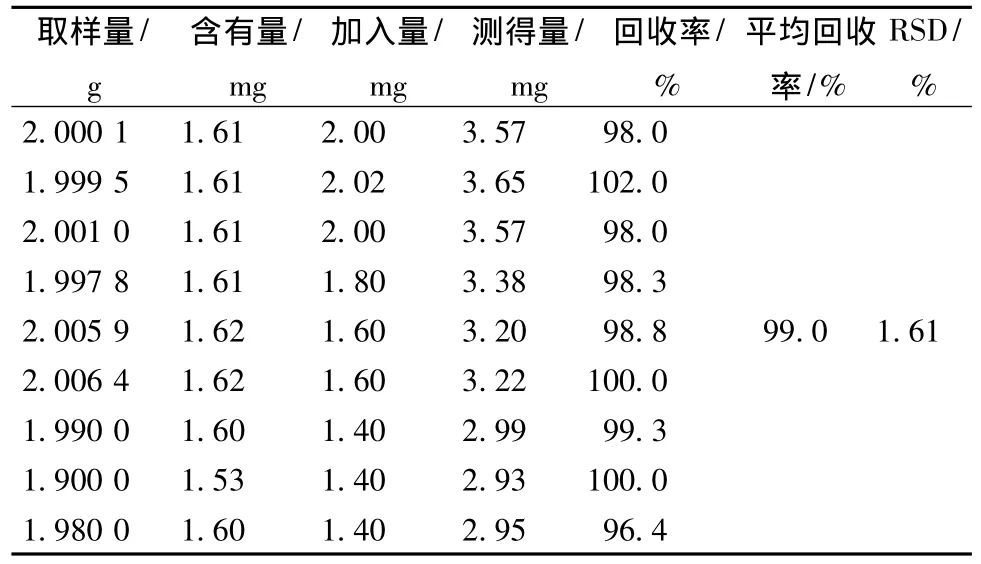
表1 加样回收试验结果 (n=9)Tab.1 Results of recovery tests(n=9)
2.2 银朱中HgS的测定 矿物药银朱在巴特日七味丸的制备过程中,部分入药,部分用于包衣,投料的比例相对较大。银朱药用时主要有安神、消炎、镇静催眠功效。银朱中汞的存在形式除硫化汞外,还有游离汞和可溶性汞盐。游离汞是0价汞,可溶性汞盐是指能溶于胃酸的汞盐。游离汞和可溶性汞盐经一定的肠道细菌生物转化后,可能与带甲基的物质反应形成甲基汞。人对甲基汞的吸收率可高达100%,甲基汞的毒性大[6]。本实验对银朱中硫化汞的定量测定方法进行了研究,通过对硫化汞的质量控制使游离汞和可溶性汞盐的量得到相对控制,保证用药的安全和有效。本实验采用硫氰酸铵滴定法测定硫化汞 (HgS)[7-8]。
2.2.1 测定方法 银朱主含硫化汞 (HgS)。参照《中国药典》2010年版一部“朱砂”项下的含量测定方法,用硫氰酸铵滴定法测定硫化汞量。
取巴特日七味丸,研细,取约1.5 g,精密称定,置250 mL凯氏烧瓶中,加硝酸50 mL,凯氏烧瓶上加漏斗,加热使硝酸呈微沸2 h,放冷,滤过,滤渣及滤纸转入原凯氏烧瓶中,加硝酸钾2.0 g,硫酸20 mL,加热微沸处理30 min,放冷,加硝酸钾2.0 g,加热微沸处理30 min使银朱完全溶解,放冷,转移至250 mL锥形瓶中,用水50 mL分次洗涤烧瓶,洗液并入溶液中,加1%高锰酸钾溶液至显粉红色,2 min内不退色,再滴加2%硫酸亚铁溶液至红色消失后,加硫酸铁铵指示液2 mL,用硫氰酸铵滴定液 (0.1 moL/L)滴定至溶液显浅橙黄色,每1 mL硫氰酸铵滴定液相当于11.63 mg的硫化汞。
2.2.2 银朱药材的测定 取银朱药材3份,各约0.1 g,精密称定,同上方法进行测定,测得平均含有量为91.75%。
2.2.3 阴性对照试验 按处方制备阴性对照品(缺银朱),称取2份,每份约1.5 g,同上方法进行测定。结果显示,滴加半滴硫氰酸铵滴定液(0.1 mol/L)即显示到达滴定终点,并且试验现象明显,说明处方中的其他组分对银朱的测定没有干扰。
2.3 样品测定 取不同厂家、不同批次的供试品,按2.1.2.1项方法制备溶液,按2.1.1项下方法操作,记录色谱图,计算巴特日七味丸中大叶茜草素量,按2.2.1项测定7个批次巴特日七味丸中硫化汞的量,测定结果见表2。

表2 样品测定结果 (n=3)Tab.2 Results of content determination of samples(n=3)
结果显示,不同来源的样品以及同一来源的不同批次样品中大叶茜草素的量有变化,在0.530 mg/g~0.819 mg/g之间。巴特日七味丸中含银朱以硫化汞计,含有量在27.18~45.23 mg/g之间。银朱药材含有毒成分游离汞和可溶性汞盐,通过对硫化汞的定量测定和控制可以使游离汞和可溶性汞盐得到相对控制。本实验的研究对巴特日七味丸的安全有效用药具有意义。
3 讨论
3.1 流动相的选择 按2.1.2.1项下方法制备供试品溶液,对以下3种流动相进行了比较研究[9-10]:甲醇-乙腈-0.2%磷酸溶液 (25 ∶50 ∶25)、甲醇-乙腈-水 (6∶3∶1)和甲醇-水 (85∶15)。结果表明,以甲醇-水 (85∶15)作为流动相时,大叶茜草素峰的保留时间为20.2 min,且与相邻组分峰分离效果好。
3.2 检测波长的选择 在200~800 nm范围内对大叶茜草素进行光谱扫描,结果显示,大叶茜草素在218.00、250.00、273.00、281.50、392.00 nm 处有吸收。分别将巴特日七味丸供试品溶液和大叶茜草素对照品溶液20μL注入高效液相色谱仪,对紫外检测器设定不同检测波长[11]的情况下,对大叶茜草素峰进行检测,并计算巴特日七味丸中大叶茜草素的量。结果表明,在250 nm波长处,大叶茜草素量最高,故确定250 nm作为检测波长,与《中国药典》2010年版一部茜草项下的检测波长一致。
3.3 对大叶茜草素和大叶茜草素溶液的稳定性考察 大叶茜草素在常温不避光的条件下稳定性差,在薄层色谱中可显示两个斑点。用HPLC法考察大叶茜草素甲醇溶液的稳定性,常温不避光条件下的稳定时间约为6 h[12],避光冷藏,该时间可延长到4倍以上,因此,建议将大叶茜草素对照品及溶液避光冷藏。
3.4 提取溶剂和提取时间的选择 本实验采用《中国药典》2010年版一部的茜草项下的提取方法,即超声提取。对甲醇、乙醇和80%甲醇作为溶剂的结果进行了比较研究,结果显示,用甲醇作为溶剂时的提取效率最高。
对超声时间15、30、45、60 min的结果进行了比较,结果显示,从15 min到30 min,随着提取时间的延长,提取效率增加;30 min以后,提取时间的延长使含有量有下降趋势。故将超声时间确定为30 min。
3.5 浸泡时间的选择 本实验对《中国药典》2010年一部版茜草含量测定项下的超声提取前的浸泡时间进行了筛选。设置不同的浸泡时间,考察浸泡时间对测定结果的影响。
结果显示,浸泡时间在0~5 h范围内,随浸泡时间增加,大叶茜草素含有量逐渐增大;浸泡5~8 h之间,大叶茜草素量最高;浸泡时间大于8 h后,大叶茜草素量有下降趋势,可能与大叶茜草素在甲醇溶液中的稳定性有关,故将浸泡时间规定为5~8 h。
3.6 耐用性试验 取本品,按2.1.2.1项方法制备溶液,按2.1.1项下方法操作,记录色谱图。用两个型号的色谱柱进行试验,对其结果进行对比研究,结果见表3。

表3 不同色谱柱的试验结果Tab.3 Results of different columns
结果显示,Phenomenex C18柱的峰窄而高,Diamonsil C18柱的峰宽且矮,两种柱子均能达到与相临峰的基线分离,按大叶茜草素的峰计算理论塔板数均大于10 000,两个柱子的测定结果基本一致。故实验对柱子的型号不做要求。
3.7 峰纯度检查 按2.1.2项下的方法制备供试品溶液和对照品溶液。按2.1.1项下方法操作,仅对流动相比例进行调整,分别用如下的流动相进行洗脱,甲醇-水 (85∶15)、甲醇-水 (80∶20)、甲醇-水 (78∶22)。各流动相的色谱图显示,随着流动相极性增大,大叶茜草素色谱峰的保留时间增加,但供试品溶液的主峰面积没有出现裂分现象。
进一步用高效液相色谱仪的二极管阵列检测器对被测成分大叶茜草素的峰进行纯度验证,结果表明,被测供试品溶液中大叶茜草素峰纯度为98.49%。
3.8 硝酸加热溶解时间的考察 为了保证有机破坏过程进行完全,对硝酸用量和加热溶解时间作了考察。取供试品约1.5 g,加50 mL硝酸,置凯氏烧瓶中,为避免加热过程中硝酸分解逸出过快,于凯氏烧瓶口处放一漏斗,置电炉上加热保持微沸,分别对15、30、60、90、120、150、180 min的加热时间的结果进行观察和比较。结果显示,加热时间从90 min开始,凯氏烧瓶中的棕色烟雾逐渐消失,溶液的颜色逐渐变浅;加热至120 min时,凯氏烧瓶中的棕色烟雾完全消失,溶液变得几乎无色透明;加热时间进一步延长时,观察的结果与120 min相同。故将加热时间定为120 min。
3.9 硝酸钾和硫酸作用时间、次数的考察 为保证被测成分消解完全,对硝酸钾加入次数及硫酸作用时间进行考察,经反复试验观察,结果显示,加入硫酸20 mL,硝酸钾2.0 g,加热微沸处理30 min,放冷,加硝酸钾2.0 g,加热微沸处理30 min,放冷,溶液接近无色,即消解完全。
[1]卫生部药典委员会.中华人民共和国卫生部药品标准:蒙药分册[S].1998:79.
[2]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:220.
[3]贝玉祥,郭 英,和万芳,等.诃子中总多酚含量的测定及其超声提取工艺的研究[J].生物技术,2008.18(2):82-85.
[4]唐宇伟,孙彬贤,黄德杰.参茜固经颗粒中大叶茜草素含量测定[J].中成药,2005,27(1):28-29.
[5]索菲娅,王 弘,陈世忠.不同采收期茜草中大叶茜草素含量的动态研究[J].中成药,2006,28(10):1499-1501.
[6]周超凡,林育华.传统中药朱砂应用概况及其安全性[J].药物不良反应杂志,2008,10(3):184-189.
[7]王东平,魏立新,杜玉枝,等.藏药“佐太”中硫化汞含量测定的方法学考察[J].时珍国医国药,2010,21(6):1359-1363.
[8]陈 燕,易进海,刘玉红,等.二十五味珊瑚丸中酯型生物碱和硫化汞的含量测定[J].中国实验方剂学杂志,2010,3(16):44-47.
[9]薛 丽,陈世忠,索菲娅,等.HPLC法测定茜草饮片中大叶茜草素的含量[J].药物分析杂志,2009,29(3):363-365.
[10]郭桂明,蔡 乐,梁小雨,等.HPLC法测定茜草饮片中大叶茜草素和羟基茜草素的含量[J].北京中医药,2011,30(7):541-542.
[11]张 宁,杨晓宏,马晓帆.HPLC法测定十三味菥蓂胶囊中大叶茜草素的含量[J].药物分析杂志,2007,27(9):1475-1477.
[12]刘 广,齐 娜,徐本明.大叶茜草素在甲醇溶液中的稳定性研究[J].华西药学杂志,2010,25(3):320-322.
猜你喜欢
河南大学学报(医学版)(2020年4期)2020-08-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
宝藏(2019年6期)2019-07-04
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
天然产物研究与开发(2018年10期)2018-11-06
中成药(2018年5期)2018-06-06
中国现代中药(2017年4期)2017-09-21
