
Huntington舞蹈病4个家系IT15基因突变的研究☆
2013-09-14 08:34邢世会陈玲陈曦李洵桦曾进胜黎锦如
中国神经精神疾病杂志 2013年10期
邢世会陈玲陈曦李洵桦曾进胜黎锦如
Hungtington舞蹈病(Huntington disease,HD)是一种常染色体显性遗传性神经系统变性疾病,表现为进行性的舞蹈样动作,智能减退及精神异常[1]。平均发病年龄30~50岁,病程约为10~15年。Gusellla等[2]首先将HD的致病基因定位于4号染色体。研究发现,HD由该染色体IT15基因CAG三核苷酸重复序列的异常扩展所致[3]。在我国HD的研究多为单一家系报道,本研究对中国南方地区4个HD家系的患者的临床和遗传学特征进行分析,以期进一步明确我国HD的发病特点。
1 对象与方法
1.1 研究对象 收集中山大学附属第一医院神经科就诊的四个HD家系,分别来自于中国广东省、福建省。据先证者线索,对各家系进行详细家系分析和临床资料采集。调查的4个家系中均有已发病患者及未发病亲属(见图1)。4家系73例成员参与该研究,其中患者13例(男9例,女4例;平均年龄46.85±13.10岁;平均发病年龄41.23±10.28岁),表型正常亲属60例(男43例,女34例;平均年龄33.00±6.25岁)。家系A 4代共有7例患者(3例已去世),男4例,女3例,AⅡ9是先证者,其母亲(AⅠ2)、哥哥(AⅡ2和AⅡ8)、外甥(AⅢ1、AⅢ3)和外甥女(AⅢ4)发病。家系B四代8名患者(4例去世),先证者BⅢ1,其外祖父(BⅠ1)、母亲(BⅡ1)、舅舅(BⅡ2、BⅡ3)、哥哥(BⅢ3)、妹妹(BⅢ5)、表妹(BⅢ10)均发病。家系C系具有明显的父系遗传特点,四代7例患者全部为男性(3例已死去),先证者CⅢ3,其父亲(Ⅱ1)、叔叔(Ⅱ4)、祖父(Ⅰ1)、兄(堂兄)弟(Ⅲ1、Ⅲ5、Ⅲ8)发病。家系D三代有3名患者,先证者DⅡ3,其父亲(Ⅰ1)和妹妹(Ⅱ1)发病。除AⅢ1患者首发症状为精神障碍,其他患者全部以运动障碍为首发症状。HD诊断依据主要依据病史、家族史和临床表现。

图1 HD家系系谱图
1.2 研究方法
1.2.1 静脉血DNA提取 经知情同意后,取其外周静脉血5ml(枸橼酸钠抗凝),应用DNA抽提试剂盒(Takara公司)提取白细胞中基因组DNA置于-20℃保存备用。
1.2.2 PCR扩增IT15基因CAG重复拷贝 参考文献[3]设计引物:5’-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3’和 5’-GGCGGTGGCGGCTGT TGCTGCTGCTGCTGC-3’,委托上海Songon公司合成。PCR反应体系为50µL,其中含Buffer II 25µL,dNTP 8 µL,基因组DNA 50~500 ng,引物各200ng,Takara LA Taq酶 1U(Takara公司),灭菌去离子水补充至50µL,PTC-200 DNA热循环仪进行PCR扩增,反应条件:94℃下预变性1 min;94℃30S,67℃ 30S,72℃ 1 min,循环 2 次;94℃ 30S,66℃ 30S,72℃ 1 min,循环 2 次;94℃ 30S,65℃30S,72℃ 1 min,循环 2 次;94℃ 30S,64℃ 30S,72℃ 1 min,循环30次;72℃延伸7 min。取PCR产物8µL于2%的琼脂糖凝胶中电泳,电泳结果凝胶成像系统拍照。
1.2.3 IT15基因CAG扩增产物基因测序 IT15基因CAG扩增产物经凝胶电泳后采用DNA凝胶回收试剂盒(Takara公司)进行纯化回收,纯化回收产物与pMT-18T克隆载体(Takara公司)连接,转化感受态E.coli DH5α细胞,于转化平板挑取单菌落过夜培养,提取质粒DNA委托上海博亚公司测序确定CAG重复拷贝数。
1.3 统计学方法 所有实验数据的处理均在统计软件包SPSS11.0上完成,不同性别、遗传方式间CAG重复次数的差异采用t检验,CAG重复次数与发病年龄的关系采用Pearson相关分析,P<0.05认为具有统计学显著性差异。
2 结果
2.1 IT15基因的(CAG)n动态突变检测 IT15基因CAG突变检测结果显示:4个家系中13例患者均扩增出一条正常等位基因片段(扩增DNA片段<120 bp)和一条异常等位基因片段,其泳动显著减慢,扩增DNA片段>150 bp。在家系A中一例临床表型正常亲属(III13)其中一条等位基因扩增片段<120bp,另一条等位基因扩增DNA片段>150 bp,疑为异常等位基因。其他正常表型患者亲属均扩增出一条基因片段(<120 bp)。四家系HD患者和正常表型亲属IT15基因CAG拷贝数目检测的部分结果如图2。
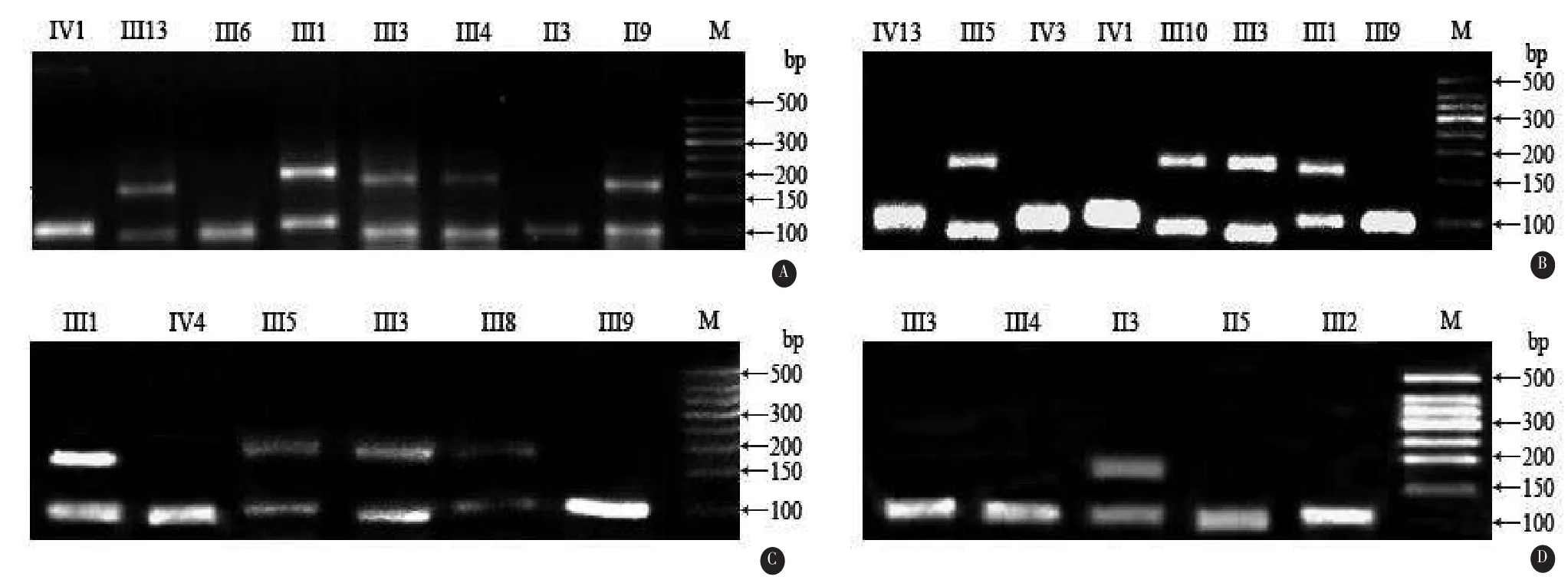
图2 四HD家系患者和部分正常表型亲属IT15基因CAG拷贝PCR凝胶电泳图
2.2 CAG重复次数在各家系中的分布 CAG扩增片段经基因测序确认其重复次数,四家系中临床确诊为HD的患者均各检测出一条异常等位基因(CAG重复次数41~51次),正常亲属中1例(AⅢ13)检出一条异常等位基因(CAG重复次数41次),确诊为症状前患者;其他亲属检测出等位基因CAG重复次数在17~20次之间。本研究对HD患者和正常表型亲属IT15基因的等位基因频率分布情况分析发现,正常亲属(除AIII13)CAG重复次数均在20次以下,其中CAG重复17次等位基因频率最高(27.59%),HD患者CAG重复48次等位基因比例最高(30.77%)。CAG重复次数与发病年龄和临床表型分布情况见表1。
2.3 CAG重复次数与临床表型关系的分析 四HD家系中所有患者CAG重复次数为45.62±2.90次,男性患者CAG重复次数为46.56±2.92次,女性患者CAG重复次数为45.75±3.30次,二者无显著性差异(P=0.26)。父系遗传的HD患者有9例,其CAG重复次数为45.00±2.12次,母系遗传的HD患者有4例,其CAG重复次数为47.00±4.24次,不同遗传方式HD患者CAG重复次数无显著性差异(P=0.38)。
本研究对CAG重复次数与发病年龄的关系分析发现,IT15基因异常等位基因CAG重复次数与发病年龄呈负相关(rPearson=-0.57,r2=0.33,P=0.03);而正常等位基因与发病年龄无相关关系(rPearson=0.32,P=0.82)(如图3)。
3 讨论
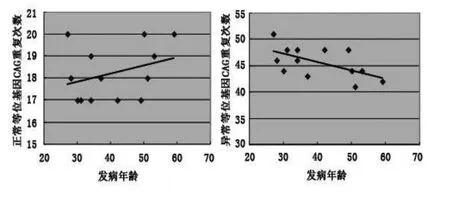
图3 HD患者IT15基因CAG重复数目与发病年龄的相关性四HD家系13例患者正常等位基因与发病年龄无相性(rPearson=0.32,P=0.82);异常等位基因CAG重复次数与发病年龄负相关(rPear-son=-0.57,r2Pearson=0.33,P=0.03)
HD是一种神经系统遗传变性疾病,不同地域、种族的人群发病率存在明显差异,欧美国家发病率较高,亚洲人群中发病率较低,中国目前尚缺乏HD发病率的流行病学资料[4]。这种发病率的差异可能与IT15基因CAG动态突变易感性有关[5-6]。依据患者典型临床表现、家族史和颅脑影像学资料(CT或MRI),多数HD患者能够得以诊断。然而,那些临床特征不典型或仅有轻微神经功能异常的患者往往经历较长时间才得以诊断[7],因此对可疑HD患者应及早进行IT15基因CAG动态突变检测。IT15基因检测具有高度敏感性和特异性,约99%的HD患者表现为CAG的异常扩展[8]。IT15基因CAG重复次数具有多态性:正常等位基因CAG重复次数多在26次以下,而异常扩展可达到36次以上,CAG重复40次以上者必然发病[9]。本研究4个HD家系IT15基因CAG突变检测结果显示,全部HD患者均携带有一个异常等位基因(CAG重复数>40次),而正常等位基因CAG重复数均在17~20次,与异常等位基因之间没有重叠。而且患者CAG重复数不同性别和遗传方式间无差异。这表明本研究中HD患者异常CAG重复次数诊断标准与国外的报道并无差异。
HD患者发病年龄呈现明显遗传早现现象,本研究中四家系系谱分析同样发现子代HD患者发病年龄较其上一代明显提前。研究已证实,HD患者的发病年龄与CAG重复次数存在负相关关系,即CAG重复次数越多,其发病年龄将会越早[10]。本研究结果显示,4家系HD患者的发病年龄与CAG重复次数之间存在负相关关系,进一步证实HD患者遗传早现规律。在四家系中有1例未发病亲属(AIII13)携带有一条异常等位基因,CAG重复数41次,当时年龄38岁,但无任何神经系统异常症状和体征,考虑为临床症状前患者。尽管HD是一种显性神经系统遗传病,然而仅50%~70%HD患者符合CAG重复次数与发病年龄负相关这一规律,其他变异情况可能与遗传变异或环境因素有关[11]。因此,尚不能排除延迟发病的可能,其发病年龄有待追踪观察,随着年龄增长有可能逐渐出现临床症状。理论上,HD患者中男/女性的患病率相等,但我们分析的四家系中发病率男性(72%)高于女性(28%),可能与样本数量较少有关。
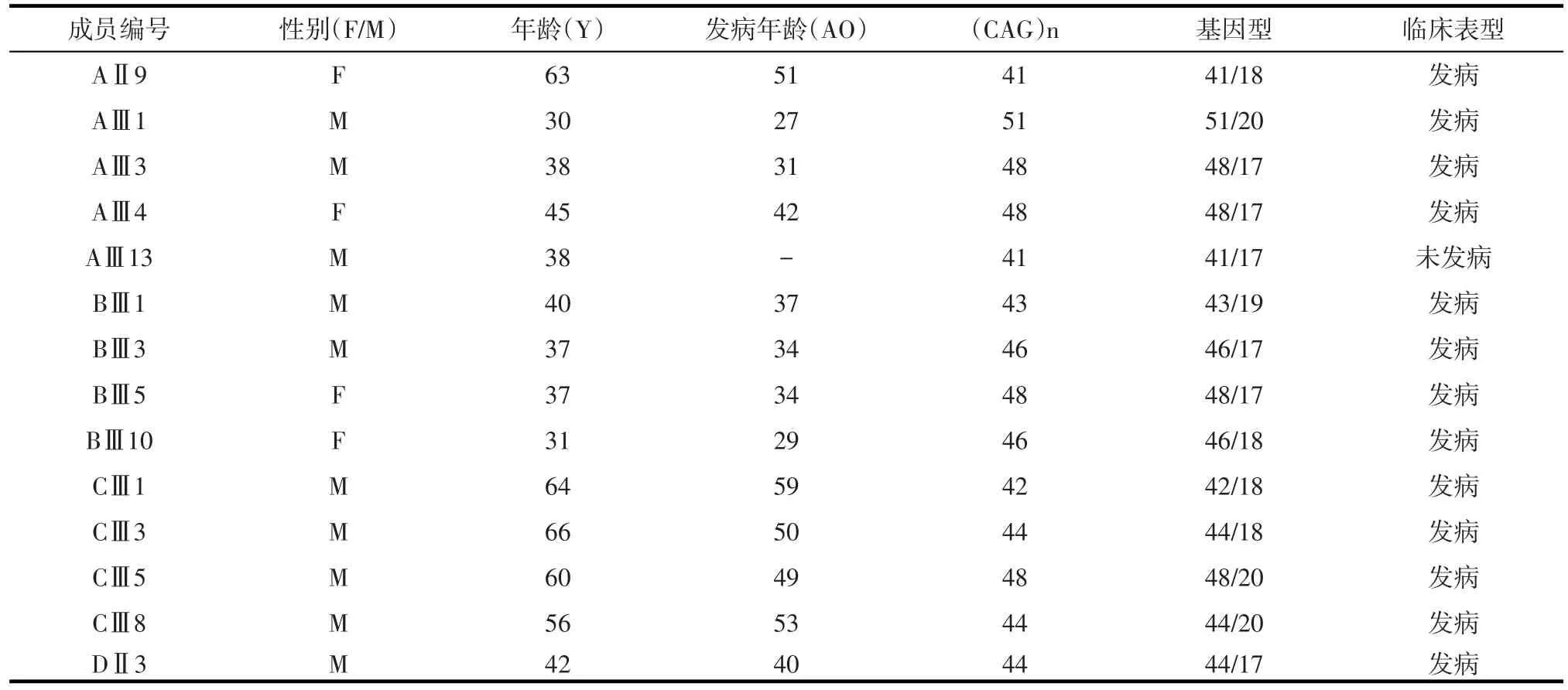
表1 4个HD家系患者及基因型与临床表型和发病年龄关系
迄今为止,HD尚缺乏有效治疗方法,避免携带异常等位基因后代出生是目前唯一手段。本实验采用PCR扩增结合基因测序方法提高了准确性和特异性。临床疑似HD或家系高危个体应及早进行IT15基因突变检测以明确诊断,从而为HD家系个体提供可靠的遗传咨询资料,并能够为产前诊断提供科学方法。
[1]Walker FO.Huntington’s disease[J].Lancet,2007,369(9557):218-228.
[2]Gusella J F,Wexler NS,Conneally PM,et al.A polymorphic DNA marker genetically linked to Huntington’s disease[J].Nature,1983,306(5940):234-238.
[3]HD Collaborative Group.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes[J].Cell,1993,72(6):971-983.
[4]Squitieri F,Andrew SE,Goldberg YP,et al.DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence[J].Hum Mol Genet,1994,3(12):2103-2114.
[5]Ma M,Yang Y,Shang H,et al.Evidence for a predisposing background for CAG expansion leading to HTT mutation in a Chinese population[J].J Neurol Sci,2010,298(1-2):57-60.
[6]Warby SC,Montpetit A,Hayden AR,et al.CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup[J]Am J Hum Genet,2009,84(3):351-366.
[7]Tabrizi SJ,Langbehn DR,Leavitt BR,et al.Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study:cross-sectional analysis of baseline data[J].Lancet Neurol,2009,8(9):791-801.
[8]Kremer B,Goldberg P,Andrew SE,et al.A worldwide study of the Huntington’s disease mutation.The sensitivity and specificity of measuring CAG repeats[J].N Engl J Med,1994,330(20):1401-1406.
[9]Almqvist E,Andrew S,Theilmann S,et al.Geographical distribution of haplotypes in Swedish families with Huntington's disease[J].Hum Genet,1994,94(2):124-128.
[10]Langbehn DR,Brinkman RR,Falush D,et al.A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length[J].Clin Genet,2004,65(4):267-277.
[11]Wexler NS.Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset[J].Proc Natl Acad Sci USA,2004,101(10):3498-3503.
猜你喜欢
广西林业科学(2022年6期)2023-01-16
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
川北医学院学报(2022年6期)2022-06-24
智慧健康(2021年17期)2021-07-30
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
遵义医科大学学报(2020年6期)2020-02-05
暨南学报(哲学社会科学版)(2015年7期)2015-11-14
作文·初中版(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
